

Contents
Whole Genome Sequence Data Types & Sizes
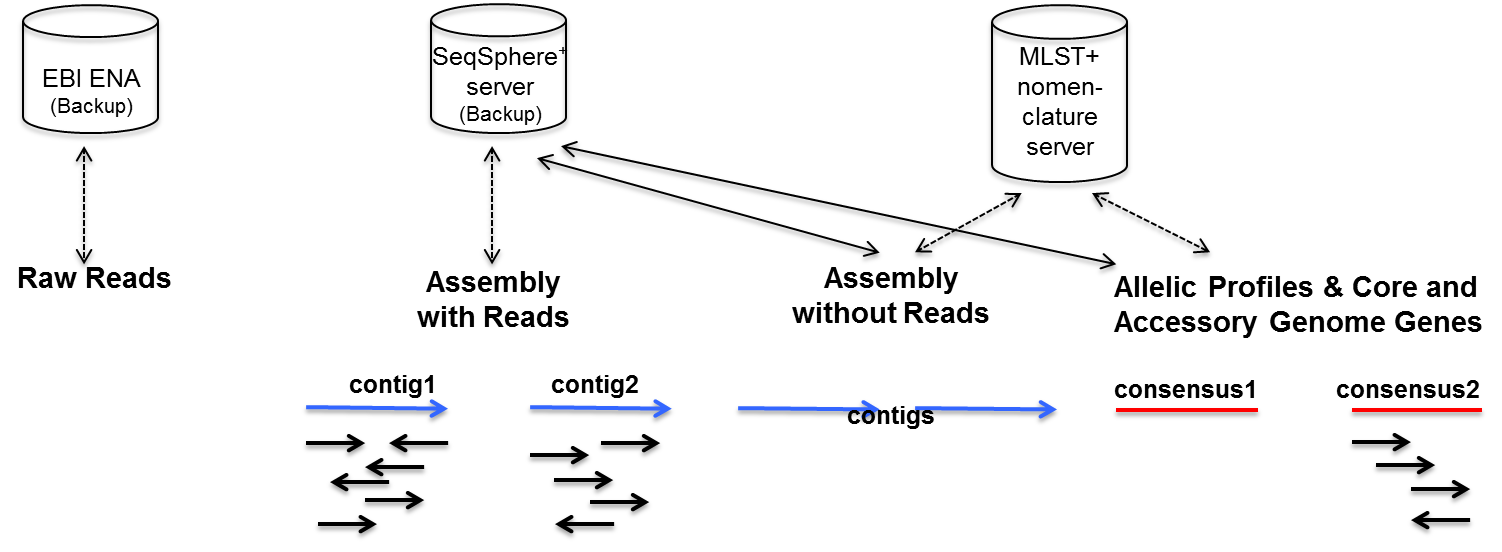
- Raw Reads
- ≈ 1 GB1) per WGS (approx. 1,000 samples per 1 TB HD; FASTQ format)
- needed for ENA submission (e.g., publication) and different assembly strategy; SeqSphere+ only knows file location if processed with pipeline, reads must be stored on a network drive to be accessible for all client computers.
- Assembly with Reads
- ≈ 200 MB1) 2) per assembly (approx. 5,000 samples per 1 TB HD; ACE or BAM format)
- needed for reproducing all SeqSphere+ results achieved with auto-correction, manual edits, and/or when dealing with multi-copy genes; optional stored and managed by SeqSphere+ on the server (may be different HD than DB HD).
- Assembly without Reads
- ≈ 1 MB per assembly (approx. 1,000,000 samples per 1 TB HD; FASTA format)
- needed for unique PCR signature extraction and for reproducing all SeqSphere+ results that does not involve auto-correction, manual edits, and/or deals with multi-copy genes; obligate stored and managed by SeqSphere+ on the server (always stored together with allelic profiles)
- Allelic Profiles & Core and Accessory Genome Genes
- ≈ 4 MB1) per assembly (approx. 250,000 samples per 1 TB HD; internal DB format)
- needed for quick search of related genomes and storing the SeqSphere+ analysis results; obligate stored and managed by SeqSphere+ on the server.
1) depending on genome size and coverage (here 5 MB & 180x fold), 2) BAM file is more than twice as large as ACE file made from same reads with same coverage.
Disk Storage Required
It is assumed that approx. 5 MB per sample (1 MB FASTA and 4 MB allelic profiles) are needed in the database directory if the sample ACE or BAM files are not stored. With the backup default settings (compress non-blocking, automatic daily backups, and keep only the last two backup files) additionally approx. 2.5 MB per sample are required for each finished backup of the database in the backup directory. Once the third backup was successfully finished the oldest backup is deleted. In addition another approx. 5 MB is temporarily needed during backup with default settings. Therefore approx. 12.5 MB (3*2.5 + 5 MB) per sample are required in the backup directory with default settings.
By default the database directory and the backup directory are located on the same hard disk of the server. However, all directory locations can be changed in the backup settings. The database directory must be on an internal hard disk. It is highly recommended to put the backup directory on a hard disk that is a different from than the one used to store the database! For performance reasons, an internal hard disk is strongly recommended.
If the backup directory is located on a network device (e.g., NAS) and/or to save some space in the backup directory, the option Create backup first in backup temp directory and move then to backup directory should be enabled. The backup is then first created in the defined backup temp directory and then moved at once to the final location. The backup temp directory must be located on an internal hard disk, ideally on a hard disk that is a different from than the one used to store the database, and requires with default settings approx. 7.5 MB (2.5 + 5 MB) per sample. The size of the backup directory reduces then to 7.5 MB (3*2.5 MB) per sample.
For example a database with 200,000 samples requires (without FASTQ/ACE/BAM files) approx. 1 TB for the database directory (on an internal hard disk) and approx. 2.5 TB for the backup directory (on a second internal hard disk; for every additional kept backup file the size increase approx. 0.5 TB).
If the option Create backup first in backup temp directory and move then to backup directory is turned on, the 200,000 samples require for backup approx. 1.5 TB in the backup directory (e.g., on a NAS or another internal hard disk; for every additional kept backup file the size increase approx. 0.5 TB) and approx. 1.5 TB in the backup temp directory (on a second internal hard disk). Your current database, backup file, and backup temporary sizes are shown in the Server Database Backup dialog on the top.
Data Size per Sample
The mean values of storage required per sample in the database are shown in the table below. The storage values are averaged for in total 14 Illumina Nextera XT library read pairs (FASTQ; 150bp, 250bp, and 300bp) from various species (C. jejuni ATCC 700819, 1.6 MBases; E. faecium TEX16, 2.7MBases; S. aureus COL, 2.8 Mbases; E. coli Sakai, 5.5 MBases; and P. aeruginosa PAO1, 6.2 MBases) sequenced with a MiSeq, de novo assembled with Velvet using default SeqSphere+ client pipeline settings, and then cgMLST & accessory target calling with SeqSphere+ using default options.
| Input data type | Data size per sample without assembly files1) | Data size per sample including assembly (ACE) files2) | Backup size per sample without/with assembly files3) |
|---|---|---|---|
| Contigs4) only (from FASTA files) | 2.4 MB | n/a5) | 1.6 MB / n/a5) |
| Raw reads (from FASTQ files6)), downsampled to 60x coverage | 2.8 MB | 73 MB | 2.2 MB / 72 MB |
| Raw reads (from FASTQ files6)), downsampled to 120x coverage | 3.6 MB | 138 MB | 2.6 MB / 137 MB |
1) always including the assembly contigs; 2)storage of assembly files (ACE/BAM) on the Server is an optional feature of SeqSphere+; 3) backing-up the database is also an optional feature of the software (can be done with or without assembly files); 4) contigs derived from 120x coverage assemblies; 5) not applicable; 6) FASTQs on average 0.65 GB large per sample. N.B.: due to less good possibility of compression a BAM file (contains all the read base quality values) is more than twice as large as an ACE file (only contains QVs for the consensus sequence) made from the same reads with the same coverage.
As raw reads (FASTQ) are the largest files of the whole workflow, it will most probably be impossible to keep them forever locally in a continuous productive setting. Raw reads are never stored on the server. Only the file location is stored (‘linked’ files). However, the raw reads can be submitted via SeqSphere+ to the European Nucleotide Archive (ENA).
ACE/BAM files can be optionally stored on the server (‘uploaded’ files). In contrast to ‘linked’ files, ‘uploaded’ files can be backuped by SeqSphere+. To make ‘linked’ files accessible from all client-computers, it is recommended to store those files on a network-drive that is assigned to all client computers with an identical path (e.g., mapping the network-drive always to the same drive letter on different Windows computers).
De Novo Assembling Runtime & Memory
Example Velvet (v1.1.04) runtimes (including AMOS to ACE file conversion) and memory requirements for Illumina Nextera XT read pairs (FASTQ) on an Intel i7-3770 system with 4 cores and 32 GB memory (purchased end of 2012 for about 1.000€) using default SeqSphere+ client pipeline quality trimming, automatic k-mer optimization, and four simultaneous Velvet processes each with 8 GB RAM allocated are shown in the table below.
| Species | Genome size | Coverage | Read length | Runtime Velvet | Used memory for single process | Used memory total |
|---|---|---|---|---|---|---|
| S. aureus COL | 2.8 MBases | 131x | 150bp PE | 15 min | ~1 GB | ~4 GB |
| S. aureus COL | 2.8 MBases | 150x | 250bp PE | 21 min | ~1.6 GB | ~6.5 GB |
| E. coli Sakai | 5.5 MBases | 150x | 150bp PE | 22 min | ~2 GB | ~8 GB |
| E. coli Sakai | 5.5 MBases | 150x | 250bp PE | 43 min | ~5 GB | ~20 GB |
| P. aeruginosa PAO1 | 6.2 MBases | 67x | 150bp PE | 18 min | ~2 GB | ~8 GB |
| P. aeruginosa PAO1 | 6.2 MBases | 150x | 250bp PE | 66 min | ~8 GB | ~32 GB |
Note that actual time and memory requirements may be different depending on genome size, read length, coverage, read distribution, sequencing quality, and k-mer settings.
The Velvet version employed is not multi-threading capable. However, more than one instance/process of Velvet can be run in parallel. Usually the available memory is the limiting resource for the maximum number of parallel Velvet processes possible. In addition, the maximum number of parallel processes possible is limited by the number of ‘virtual cores’ available (for most modern processors, e.g. i5 or i7, this is usually the number of cores available times two). In pipeline mode one sample after another is sequentially worked-up.
The above table helps to estimate how much memory is needed and thereby how many processes of Velvet can be run in parallel. For example, on a system with 32 GB RAM, up to 4 processes can be run in parallel for a genome size of up to 6.2 MBase using down-sampling to 150x coverage and default trimming (assuming that the processor supports so many parallel processes). For smaller genomes or less coverage, a higher number of processes can be run in parallel if enough processor ‘virtual cores’ are available. By default (configurable) SeqSphere+ starts one Velvet process per 8 GB of RAM available on the client-computer. If a memory error should occur then this failure is documented expressively in the log-files.
De novo assembly is the speed limiting step of the whole cgMLST analysis procedure when done from raw reads (scanning for core/accessory genome genes with SeqSphere+ usually takes less than 5 min).
Reference Mapping Runtime
Example runtimes of BWA (-sw, v0.6.1-r104) mapping (including the HD intensive conversion of SAM to sorted BAM file) and combined runtimes of BWA and SeqSphere+ cgMLST & accessory target calling for Illumina Nextera XT read pairs (FASTQ) of M. tuberculosis H37Rv (4.4 MBases) on an Intel i7-3770 system with 4 cores and 32 GB memory (purchased end of 2012 for about 1.000€) using default SeqSphere+ client pipeline quality trimming, downsampling, and BWA assembly with a maximum of eight simultaneous threads are shown in the table below.
| Coverage | Runtime BWA 150bp PE | Runtime combined 150bp PE | Runtime BWA 250bp PE | Runtime combined 250bp PE |
|---|---|---|---|---|
| 10x | 2 min | 5 min | 3 min | 6 min |
| 20x | 3 min | 5 min | 4 min | 7 min |
| 30x | 3 min | 6 min | 4 min | 7 min |
| 40x | 4 min | 7 min | 5 min | 8 min |
| 50x | 5 min | 8 min | 6 min | 8 min |
| 60x | 5 min | 8 min | 6 min | 9 min |
| 70x | 6 min | 9 min | 7 min | 10 min |
| 80x | 6 min | 10 min | 7 min | 10 min |
| 90x | - | - | 8 min | 11 min |
| 100x | - | - | 8 min | 12 min |
| 120x | - | - | 9 min | 12 min |
Note that actual time may be different depending on genome size, read length, coverage, read distribution, and sequencing quality.
The BWA version employed is multi-threading capable. The maximum number of parallel threads possible is limited by the number of ‘virtual cores’ available (for most modern processors, e.g. i5 or i7, this is usually the number of cores available times two). In addition to the available ‘virtual cores’ the available memory may be limiting. However, reference mapping is not very much memory demanding. 4 GB RAM in total available is usually more than enough. Therefore, if at all the number of ‘virtual cores’ available is the limiting resource for the maximum number of parallel BWA threads possible. By default (configurable) SeqSphere+ starts BWA with a maximum number of threads equaling the number of ‘virtual cores’ available on the client-computer. In pipeline mode one sample after another is sequentially worked-up.
Reference mapping is far less time and computationally expensive than de novo assembly.
Assembling, Genome-wide Target Scan, and Allele Calling Runtime
Example runtimes of Velvet (v1.1.04) or BWA (-sw, v0.6.1-r104) assembly and cgMLST & accessory target scanning with SeqSphere+ for Illumina Nextera XT read pairs (FASTQ) on an Intel i7-3770 system with 4 cores and 32 GB memory (purchased end of 2012 for about 1.000€) using default SeqSphere+ client pipeline quality trimming, downsampling, and for Velvet assembly automatic k-mer optimization and four simultaneous processes each with 8 GB RAM allocated or for BWA assembly with a maximum of eight simultaneous threads are shown in the table below.
| Species | Genome size | cgMLST / Accessory targets | Assembly | Read length | Runtime genome-wide target scan and allele calling | Runtime 60x coverage assembly and genome-wide target scan and allele calling | Runtime 120x coverage assembly and genome-wide target scan and allele calling |
|---|---|---|---|---|---|---|---|
| C. jejuni ATCC 700819 | 1.6 MBases | 637 / 958 | de novo (Velvet) | 150bp PE | 1 min | 6 min | 10 min |
| E. faecium TEX16 | 2.7 MBases | 2,018 / 317 | de novo (Velvet) | 150bp PE | 1 min | 10 min | 16 min |
| 250bp PE | 1 min | 14 min | 22 min | ||||
| S. aureus COL | 2.8 MBases | 1,861 / 706 | de novo (Velvet) | 150bp PE | 1 min | 11 min | 17 min |
| 250bp PE | 1 min | 13 min | 21 min | ||||
| E. coli Sakai | 5.5 MBases | 4,333 / 318 | de novo (Velvet) | 150bp PE | 2 min | 20 min | - |
| 250bp PE | 1 min | 21 min | 39 min | ||||
| P. aeruginosa PAO1 | 6.2 MBases | 3,842 / 1,670 | de novo (Velvet) | 250bp PE | 6 min | 32 min | 56 min |
| M. tuberculosis H37Rv | 4.4 MBases | 3,257 / 438 | mapping (BWA) | 150bp PE | 1 min | 8 min | - |
| 250bp PE | 1 min | 9 min | 12 min |
The whole assembly and cgMLST allele calling procedure usually takes less than 1h per sample (if processed with recommended maximum coverage of 120x). Therefore, one SeqSphere+ client easily scales with the output of one MiSeq machine run in 24/7 mode.