

Ridom Typer can be used to download FASTQ files from NCBI Sequence Read Archive (SRA).
Invoke the function Tools | Download FASTQ from SRA to open a dialog window and enter or import the NCBI accessions that should be downloaded.
The following types of accessions are supported (NCBI, EMBL-EBI, DDBJ):
- SRA Run Accession (
SRR.., ERR..., DRR...) - SRA Experiment Accession (
SRX.., ERX..., DRX...) - SRA Sample Accession (
SRS.., ERS..., DRS...) - SRA Study Accession (
SRP.., ERP..., DRP...) - BioSample Accession (
SAM...) - BioProject Accession (
PRJ...)
(See more details at NCBI)
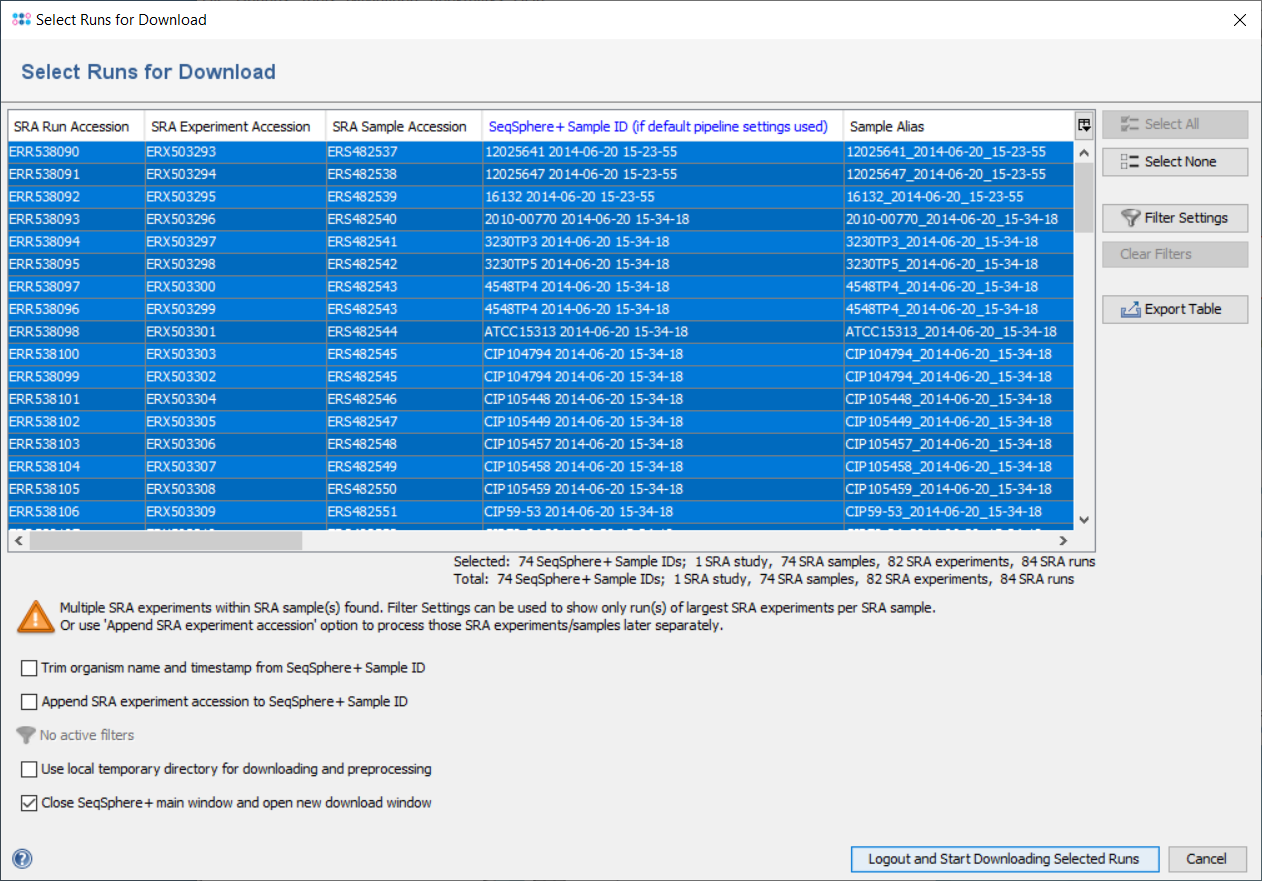
The found metadata for the given accessions are shown in a table, where each row represents one SRA run. Each SRA run belongs to a SRA experiment, each SRA experiment belongs to a SRA sample, and each SRA sample belongs to a SRA study. A SRA experiment can contain multiple SRA runs done from the same library. A SRA sample can contain multiple SRA experiments and it is usually not a good idea to assemble reads across various experiments. All SRA samples have a Sample Alias and most SRA samples have a Strain Name and a Sample Title that all must not be unique. By default the Strain Name is taken as Ridom Typer Sample ID and the FASTQ File Name Trunk. If the SRA sample has no Strain Name attached then the Sample Alias or the Sample Title is taken instead. The underscore and other special characters (e.g., !, :, /) are in the Ridom Typer Sample ID and the FASTQ File Name Trunk replaced against empty space (the unchanged names are stored in the searchable Strain and Alias ID(s) Ridom Typer data fields, respectively). In addition the SRA run accession is attached with a leading underscore to the FASTQ File Name Trunk.
The Ridom Typer Sample ID determines how the downloaded reads are treated. If there are potential problems with the Sample ID, context-sensitive warnings are shown below the table in the left corner of the window. Downloading FASTQs and metadata with default settings would result in assembling multiple SRA runs of the same SRA experiment together once a pipeline with default file naming parameters would be started. However, also multiple SRA experiments of the same SRA sample would be assembled together. To avoid this latter case either use the 'Append SRA experiment accession to Ridom Typer Sample ID' checkbox option to assemble the experiments separately or apply the 'SRA experiments' filter. Similar, if there would be SRA samples with the same Strain Name also those reads would assemble wrongly together. In this case either use the the 'Append SRA experiment accession to Ridom Typer Sample ID' checkbox option to assemble the SRA samples separately or apply the 'SRA samples' filter. Finally, in the unlikely case that two SRA samples have no Strain Name but the same Sample Alias, a warning 'Different SRA samples with the same Ridom Typer Sample ID were found!' would be shown. Again the issue could be solved by using the 'Append SRA experiment accession to Ridom Typer Sample ID' checkbox option to assemble the SRA samples separately.
The table can be filtered using the button Filter Settings. Four different filters are available:
- The runs can be filtered by a sequencer vendor, e.g. ILLUMINA (called "sequencer platform" in SRA).
- The runs can be filtered by the sequencing protocol, i.e. PAIRED or SINGLE (called "library layout" in SRA).
- SRA experiments can be filtered for SRA sample: if selected, the table shows only run(s) of the largest SRA experiment, if multiple SRA experiments from same the SRA sample are found.
- SRA samples can be filtered for strain name: if selected, the table shows only run(s) of the SRA sample with largest experiment, if multiple SRA samples are found with the same strain name.
When the FASTQ files are downloaded from SRA, Ridom Typer creates automatically a SPEC file with the same file name, that contains all available metadata. After the FASTQ files are downloaded, they can be processed using a Ridom Typer Pipeline. The metadata from SPEC files is automatically imported by the pipeline.
Ridom Typer first tries to download the SRA file via a direct https download and then creates a FASTQ file using the SRA toolkit (fastq-dump) for conversion of the file. If this approach fails for whatever reasons, then the SRA toolkit is also used to retrieve and download the FASTQ file (which takes normally longer than the direct download).
Get a List of Available Run Accessions of a Certain Species
A list of accessions for all available SRA sequences of a certain species, can be downloaded from the SRA website using the following steps:
- Step 1: Go to the SRA website https://www.ncbi.nlm.nih.gov/sra/
- Step 3: Search for genus and species name (e.g., Listeria monocytogenes).
- Step 4: Click on the left in section Source on DNA to filter the results.
- Step 5: Click on the left in section Type on genome to filter the results.
- Step 6: Click on the right in section Results by taxon / Top Organisms on the organism.
- Step 7: Click on the upper right of the result list on Send to and choose File as Destination and Accession List as Format to download a list of run accessions.
The list of run accessions can be entered in the Ridom Typer Tools | Download FASTQ from SRA dialog to download the metadata and the FASTQ files. The metadata could also be exported and a tool like MS Excel could be used to filter and/or sort (e.g., for country and/or time) the run accessions. The remaining run accessions could then be entered again in the Ridom Typer Tools | Download FASTQ from SRA dialog to download and process only the data of the samples required for analysis.
FOR RESEARCH USE ONLY. NOT FOR USE IN CLINICAL DIAGNOSTIC PROCEDURES.