

Overview
FastQC (citation) is distributed with Ridom Typer and can be used to perform the following quality checks for FASTQ or SAM/BAM files:
- Per base sequence quality
- Per tile sequence quality
- Per sequence quality scores
- Per base sequence content
- Per sequence GC content
- Per base N content
- Sequence Length Distribution
- Sequence Duplication Levels
- Overrepresented sequences
- Adapter Content
The checks are resulting in succeeded, warning, or failed according to predefined thresholds.
Perform Read Data Quality and Adapter Control in Pipeline
If the General Settings option Perform read data quality and adapter control (FastQC) is enabled in the pipeline script, Ridom Typer starts FastQC for all FASTQ files that are processed.
As not all checks done by FastQC are of relevance for general sequence quality problems, only two of them are evaluated in the Ridom Typer pipeline:
- Per Base Sequence Quality
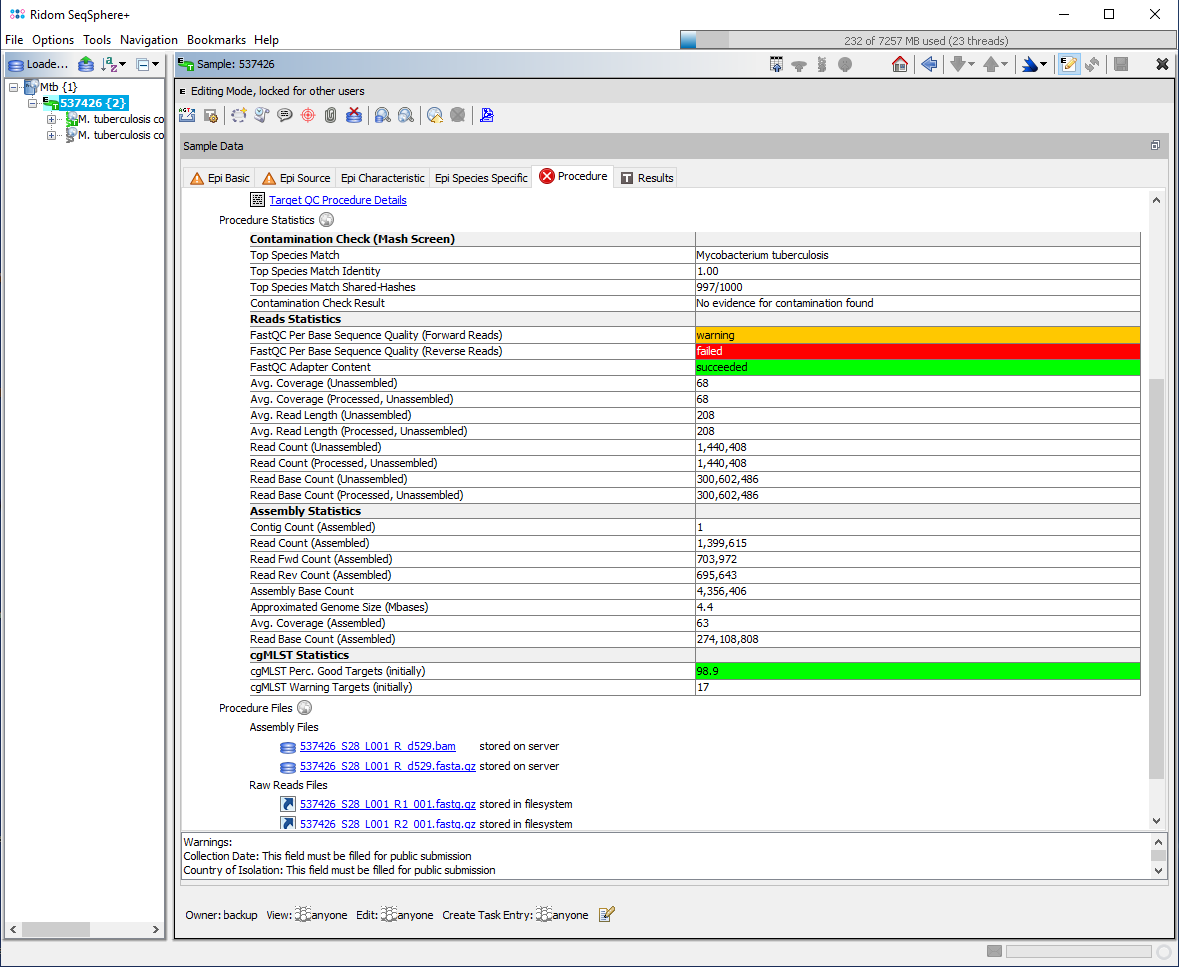
- Checks the base call qualities in the FASTQ file. If the lower quartile for any base is less than 10 or if the median for any base is less than 25 the result will be 'warning'. If the lower quartile for any base is less than 5 or if the median for any base is less than 20 the result will be 'failed'. The result is also stored in a graph, where the y-axis shows the quality scores. The quality of base calls on most sequencing platforms will degrade as the run progresses. Therefore, the quality is decreasing towards the end of a read and forward reads have usually a better quality than reverse reads. The results are stored in the procedure statistic fields FastQC Per Base Sequence Quality (Forward Reads) and FastQC Per Base Sequence Quality (Reverse Reads). Warnings for the reverse reads are ignored as they appear very often with Illumina data.
- Adapter Content
- Checks if the reads in the FASTQ file contain a significant amount of adapter sequences. If an adapter sequence is present in more than 5% of all reads the result will be 'warning'. If more than 10% are present the result will be 'failed'. The result is also stored in a graph, where the y-axis shows the amount of adapter content. If a significant amount of adapter sequences are found adapter trimming is recommended. The result is stored in the procedure statistic field FastQC Adapter Content. They are usually identical for the forward and the reverse read files.
The result fields are highlighted in the procedure tab of a loaded sample. The right-click menu in the table of the procedure tab allows to view the stored graphs for the two FastQC checks.
Tools Menu FASTQ Quality Control (FastQC)
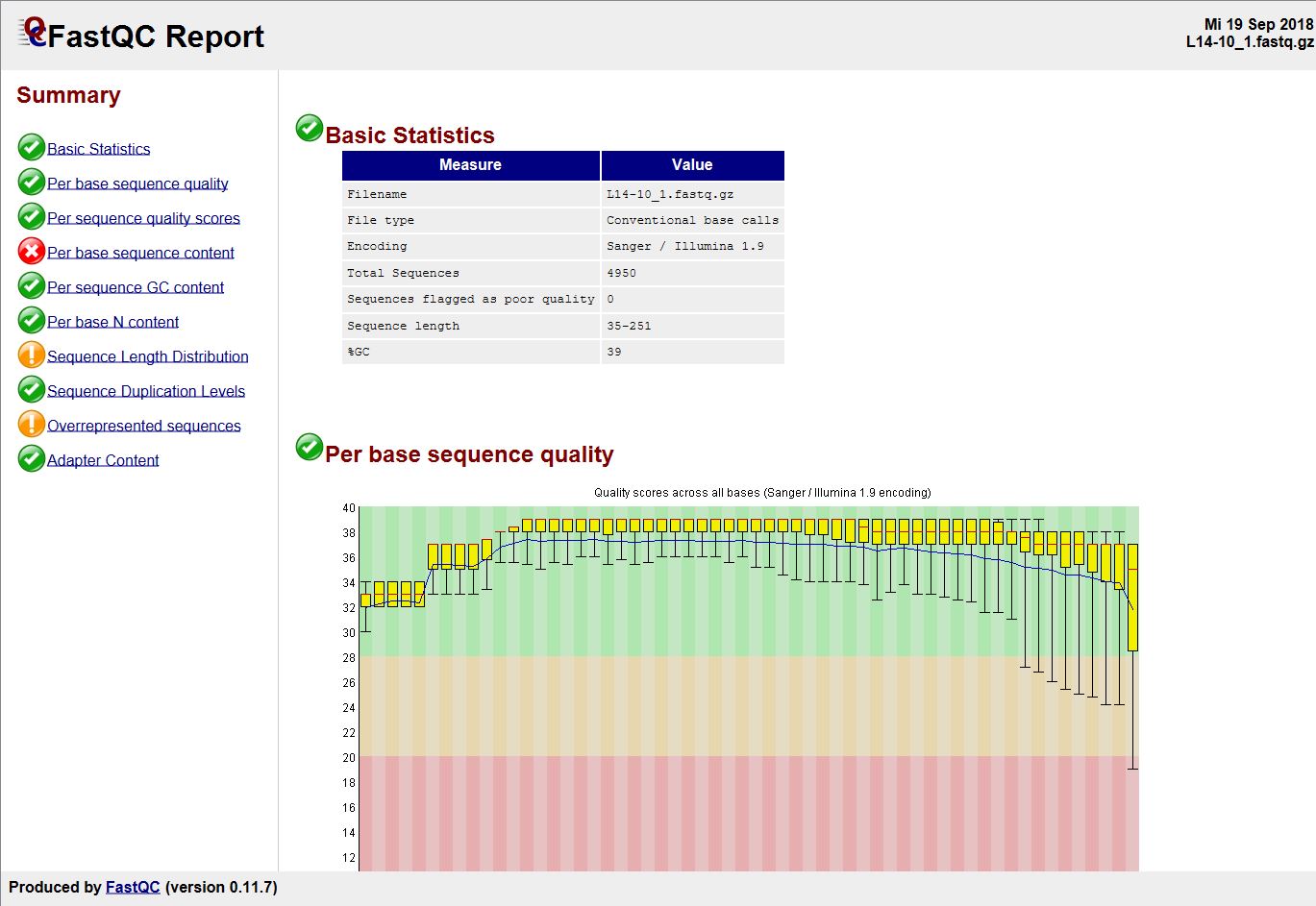
The menu function Tools | Genome Utilities | FASTQ Quality Control (FastQC) can be used to perform the FastQC quality checking on one or more FASTQ or SAM/BAM files. After the process has finished the complete FastQC reports are shown for each selected file in a new window. The reports are exportable to HTML files.