

Contents
Overview
Mash Distance (citation) is a software for fast genome distance estimation using the MinHash algorithm. In Ridom Typer Mash Distance is used for rapid species identification and automatic project choosing in the pipeline.
For this purpose, Ridom Typer comes with a Mash reference database (sketch size of s=1,000 and k=21) that contains all prokaryotic NCBI RefSeq Genome entries with status complete or chromosome that were filtered for taxonomic reliable genus and species information (30,547 genomes, as of September 2023).
![]() Important: Mash Distance requires Linux or Windows with installed Windows Subsystem For Linux.
Important: Mash Distance requires Linux or Windows with installed Windows Subsystem For Linux.
Pipeline Automatic Project Choosing (Mash Distance)
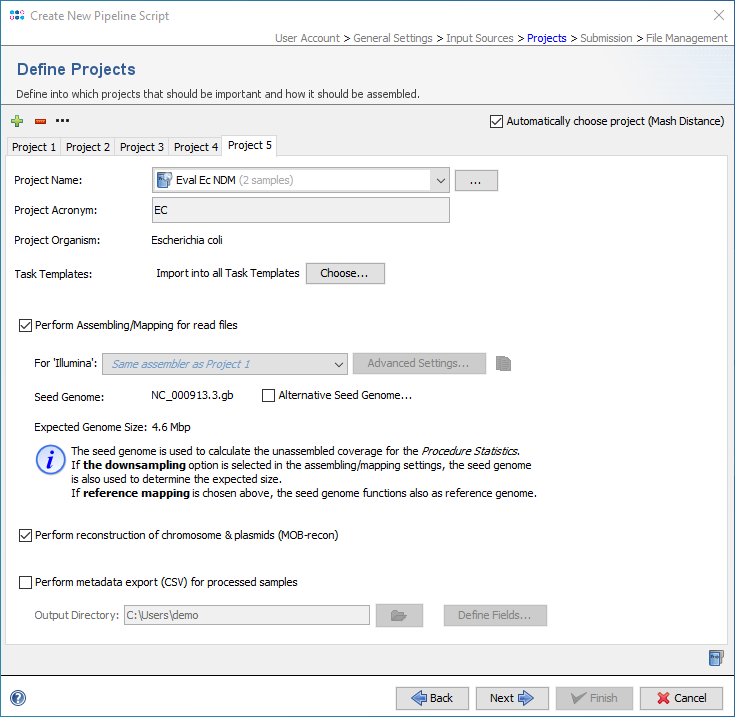
In the section Define Projects of a pipeline script, the option Automatically choose project (Mash Distance) can be enabled to choose the correct project for a processed sample automatically. If enabled, Mash Distance is started to find the closest match for the input file of a sample (FASTA/GB/BAM/ACE or first FASTQ file) in the Mash reference database. If a match above the thresholds (Mash-distance <=0.1, Matching-hashes >=100) is found, the project that provides task templates for that species is automatically chosen.
This option can only be used if only one project per species is defined in the pipeline. If no matches above the threshold were found or if no project was found in the pipeline script for a matching species, the sample processing fails and the pipeline continues with the next sample.
To be able to handle closely related and/or synonymous species, Mash equivalency groups have been defined for some species.
Distinction between Escherichia and Shigella
If the Top Match of the Mash species identification is Escherichia or Shigella, the function will try to further distinguish between these closely related genera using the presence of ipaH and Shigella-specific STs as defined by ShigaPass [PubMed 36951906]. If the genus cannot be determined this way, the species will be assigned based on the If species identification fails settings in the pipeline script.
Tools Menu Identification (Mash Distance)
.JPG)
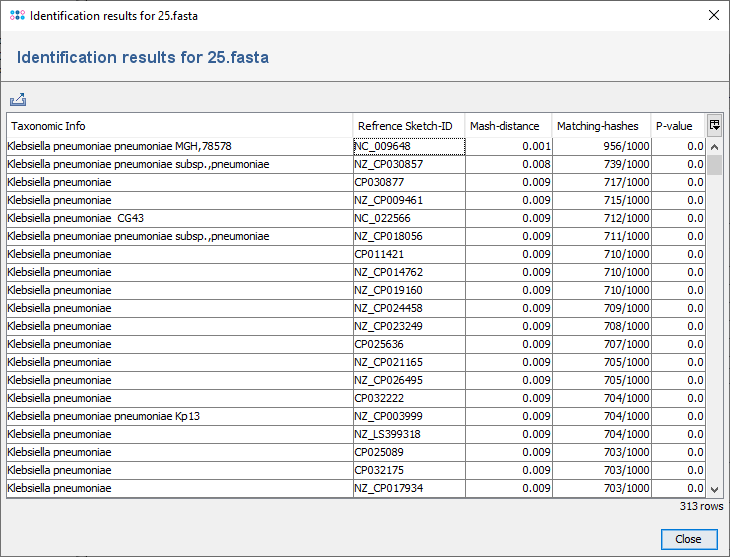
The menu function Tools | Genome Utilities | FASTQ Species ID (Mash Distance) can be used for rapid identification of a read file (FASTQ) or an assembly contigs file (FASTA/GB/BAM/ACE). For read data, the forward reads file is recommended to be used.
When the dialog is confirmed with Start, Mash Distance is started to find matches for the query in the Mash reference database. The resulting matches are filtered by thresholds for Mash-distance and Matching-hashes. The default thresholds (Mash-distance <=0.1, Matching-hashes >=100) can be changed.
The result is shown in a dialog window containing an exportable table with all matches above the defined thresholds. By selecting a row and right-clicking the entry can be browsed at the NCBI website. The table has the following columns:
- Taxonomic Info
- Contains genus, species, sub-species, and strain names of found genomes in Mash reference database.
- Reference Sketch-ID
- The NCBI accession number of found genomes in the Mash reference database. The table context menu (right-click) allows to open a web browser for the selected NCBI accession(s).
- Mash-distance
- Computed estimation of the mutation rate between two sequences directly from their internally created MinHash sketches. Distances are most accurate if calculated from assembled contigs. If calculated from FASTQ files the distances increase with increasing coverage due to accumulating sequencing errors. The Mash distances (D) correlate well with the average nucleotide identity (ANI; a common taxonomic measure of genome similarity) with D ≈ 1 - ANI.
- Matching-hashes
- The more similar two genomes are the more MinHashes are likely to match.
- P-value
- Since MinHash distances are probabilistic estimates it is important to consider the probability of seeing a given distance by chance. Lower p-values correspond to more confident distance estimates and will often be rounded down to 0.
FOR RESEARCH USE ONLY. NOT FOR USE IN CLINICAL DIAGNOSTIC PROCEDURES.