

Contents
1 Overview
This tutorial describes how to use the Ridom Typer software to analyze outbreak strains with cgMLST.
Reference mapped Mycobacterium tuberculosis whole genome shotgun (WGS) data of 4 samples of a community outbreak (isolate1, isolate2_mother_child, isolate3_mother_child, isolate4) is analyzed with the predefined public cgMLST scheme for the M. tuberculosis complex. All 4 samples are part of a larger outbreak consisting of in total 26 isolates and all exhibiting identical IS6110 DNA fingerprint and spoligotyping patterns. The outbreak was published by T. Kohl et al. (JCM 52: 2479, 2014).
M. tuberculosis is used exemplarily for this demonstration. However, by reading this tutorial you should be able to define your own projects for all species with a public available cgMLST scheme in the Task Template Sphere. If you are analyzing a species for which no public cgMLST is available yet, please take a look at Core Genome MLST Schemes. Pre-assembled WGS data is used for this tutorial. To learn how to assemble WGS data with Ridom Typer see the Pipeline Tutorial. Please note that we currently recommend only for species of the M. tuberculosis complex doing a mapping instead of a de novo assembly as it is recommended for all other species!
Like MLST, the genome-wide gene-by-gene allele calling cgMLST approach employed here, uses alleles as the unit of comparison, rather than nucleotide sequences. In allele-based comparisons among isolates, each allelic change is counted as a single genetic event, regardless of the number of nucleotide polymorphisms involved. This provides a simple and effective correction for the fact that in many bacteria, common horizontal genetic transfer events account for many more polymorphisms among specimens than rarer point mutations. The cgMLST approach retains information at all loci and avoids the need to categorize which changes are recent point mutations and which are due to recombination. As core and accessory genome MLST schemes record the sequences of allelic variants, those schemes can also be used for sequence-based analyses (e.g., SNP calling or using concatenated sequences) when this is appropriate (adapted from: Maiden MC, Jansen van Rensburg MJ, Bray JE, Earle SG, Ford SA, Jolley KA, McCarthy ND. MLST revisited: the gene-by-gene approach to bacterial genomics. Nature Rev Microbiol. 2013, 11: 728-36 [PubMed 23979428]).
2 Preliminaries
- Installation: This tutorial requires a running Ridom Typer client and server. Start the Ridom Typer server, then start the Ridom Typer client and initialize the database. For evaluation purpose a free evaluation license can be requested.
- Tutorial Data: Download the example data archive Ridom_Typer_Examples_WGS_M_tuberculosis.zip for this tutorial, and extract the zip-file on your computer.
- The example data contains whole genome shotgun (WGS) data for 4 isolates of Mycobacterium tuberculosis. The isolates are part of retrospective study where isolates of 2150 patients were analyzed with classical genotyping (Hamburg, 15 years). All 4 isolates are identical for a certain IS6110 and Spoligo type (Haarlem lineage). 2 of the 4 isolates have an epidemiological link, the 2 others have no epidemiological link. The WGS data was reference mapped to strain H37Rv (cgMLST reference strain). The data was provided by S. Niemann et al., Research Center Borstel & R. Diel, Univ. Kiel, Germany.
3 Creating Project with DB Scheme and Tasks
- Step 1: Create a new Project with the menu: File | New | Create Project. A project is typically set-up for samples of the same species. Occasionally for very closely related species, e.g., Mycobacterium tuberculosis complex or Brucella, a project holds samples of several species. For certain applications also sub-species specific, e.g., STEC, projects are recommended.
- Step 2: Enter a name for your Project (e.g., MTBC outbreak). For the Mycobcaterium tuberculosis complex a special database scheme exists that provides some species specific database fields. Choose Mycobacterium tuberculosis/bovis/africanum in the DB Scheme field.
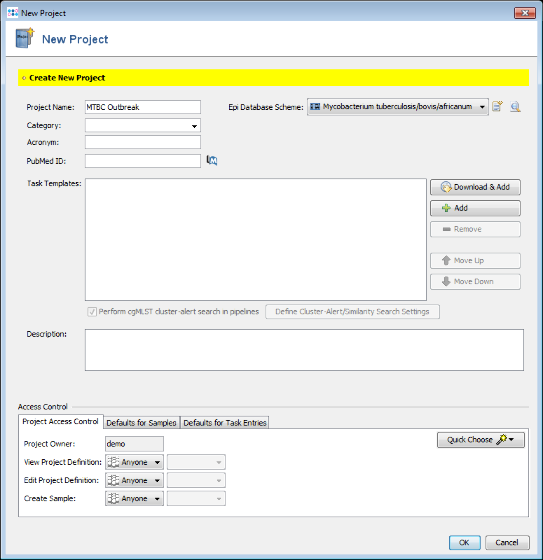
- Step 3: Press
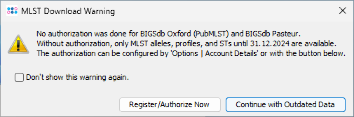
 Download & Add in Task Templates section. A MLST download warning dialog is shown. BIGSdb/pubMLST Oxford and Pasteur requires to have an account and an authorization to retrieve MLST alleles, STs, and schemes newer than 31 December 2024. For all tutorials such definitions are not needed. Therefore, just press Continue with Outdated Data to proceed. If you do not want to see this dialog again during following tutorials, mark the checkbox Don't show this warning again before clicking Continue. Once an account was registered at the pubMLST web site(s), the authorization can be activated in the Ridom Typer Account Details.
Download & Add in Task Templates section. A MLST download warning dialog is shown. BIGSdb/pubMLST Oxford and Pasteur requires to have an account and an authorization to retrieve MLST alleles, STs, and schemes newer than 31 December 2024. For all tutorials such definitions are not needed. Therefore, just press Continue with Outdated Data to proceed. If you do not want to see this dialog again during following tutorials, mark the checkbox Don't show this warning again before clicking Continue. Once an account was registered at the pubMLST web site(s), the authorization can be activated in the Ridom Typer Account Details.
- Step 4: Choose as organism Mycobcaterium tuberculosis.
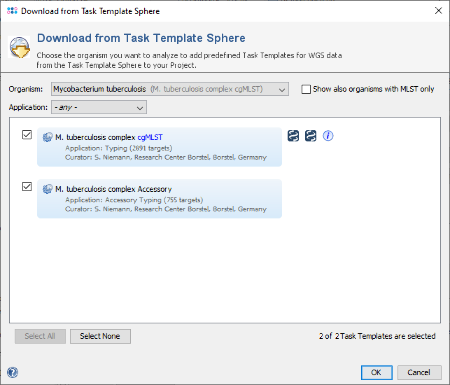
- Step 5: There are five Task Templates available for M. tuberculosis. Choose only the first two: cgMLST and Accessory. The cgMLST Task Template defines the 2891 genes of the seed genome strain H37Rv that are used for the stable typing and for the definition of the complex type. The Accessory Task Templates defines in addition 755 genes that do not belong to the core genome. However, they can be used to increase the discriminatory power if the resolution of cgMLST is not high enough. Select both Task Templates and press OK to download and to add them to the Project.
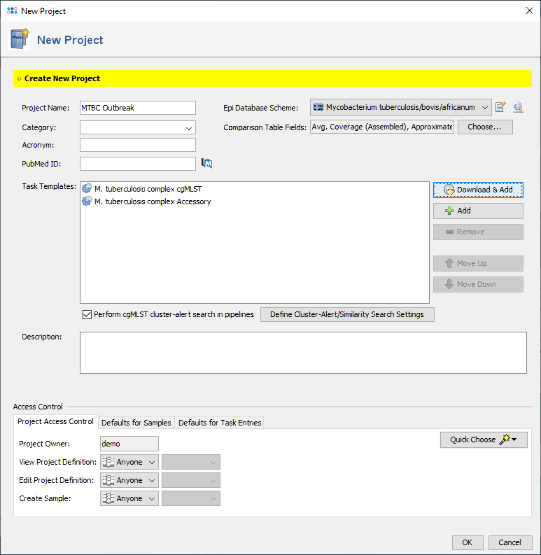
- Step 6: The Project now contains two Task Templates. Confirm with 'OK' to save the Project.
- Ridom Typer is a resequencing software. Once you have setup a project like this you can literally analyze hundreds/thousands of sequence data automatically.
4 Import Outbreak Strains to cgMLST
- Step 1: Choose from the menu File | Process Assembled Genome Data
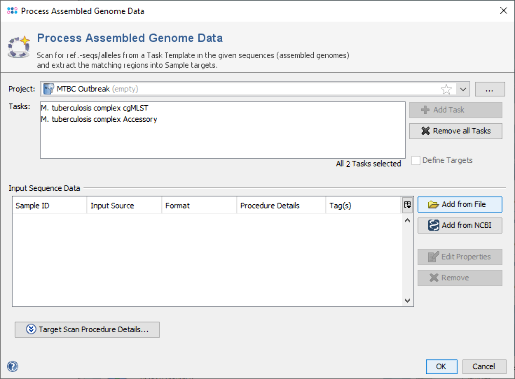
- Step 2: The new cgMLST Project you just created and both Task Templates should be preselected already. Now use the button
 Add from File and choose the 4 fasta-files from the tutorial data folder. Those files are contigs from NGS data that were reference mapped against H37Rv (NC_000962.3) which is the seed genome of the cgMLST scheme. Push the Open button.
Add from File and choose the 4 fasta-files from the tutorial data folder. Those files are contigs from NGS data that were reference mapped against H37Rv (NC_000962.3) which is the seed genome of the cgMLST scheme. Push the Open button.
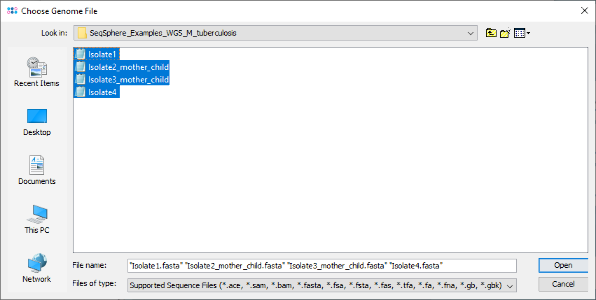
- Step 3: The upcoming window allows to specify sequencing information. Go with the default settings here by clicking OK. Confirm again with OK in the Process Assembled Genome Data window to start the import process.
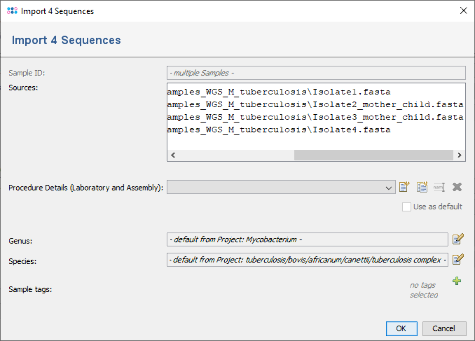
- Step 4: Ridom Typer now loads all input data and searches for the target reference sequences that are defined in the Task Template (using a built-in BLAST).
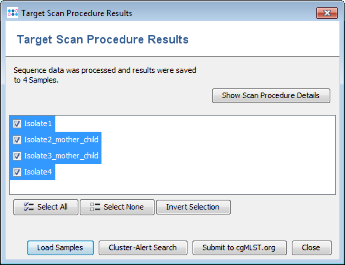
- Step 5: Once the import is completed after some minutes, a dialog window shows the imported Samples. Press Load Samples to open the newly processed Samples.
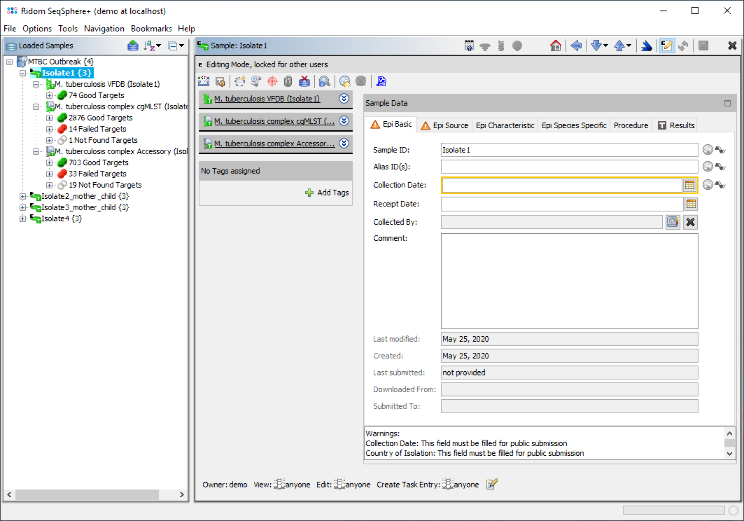
- Step 6: The upcoming navigation tree shows all new Samples. Each Sample node in the navigation has two sub nodes: The cgMLST task and the Accessory task. Below the task nodes there are the target nodes. Each target node represents one sequence (here a gene) extracted from the input data (genomes or wgs contigs). The targets can have different states:
-
 Good Targets (green) were extracted and fulfilled all requirements that are defined in the Target QC Procedure of the Task Template.
Good Targets (green) were extracted and fulfilled all requirements that are defined in the Target QC Procedure of the Task Template.
-
 Failed Targets (red) were extracted, but failed at least in one of the requirements that are defined in those parameters. For example, they may have frame shifts and incorrect lengths compared to the published H37Rv strain sequence that was used as reference.
Failed Targets (red) were extracted, but failed at least in one of the requirements that are defined in those parameters. For example, they may have frame shifts and incorrect lengths compared to the published H37Rv strain sequence that was used as reference.
-
 Not Found Targets (gray) were not extracted (because the match did not reached the thresholds in the previous step)
Not Found Targets (gray) were not extracted (because the match did not reached the thresholds in the previous step)
-
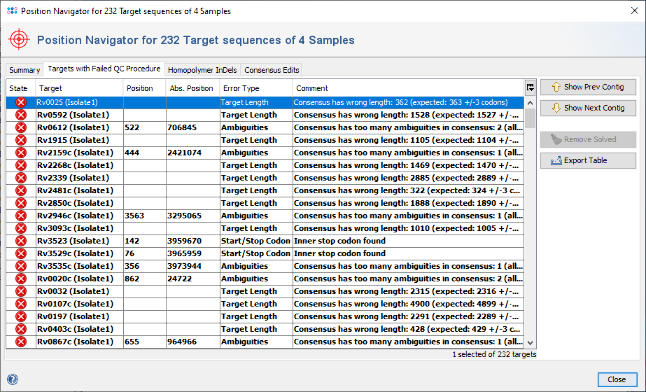
- Step 7: Select
 Tools | Position Navigator from the menu. By default all red targets are selected, so just click OK. A confirmation dialog appears. Press OK to continue. After a short while the Position Navigator is opened. Click on the tab Targets with Failed QC Procedure to see a table with the problems that appeared in the imported sequence data. You can double click on a row in the table to jump to the according position in the contig.
Tools | Position Navigator from the menu. By default all red targets are selected, so just click OK. A confirmation dialog appears. Press OK to continue. After a short while the Position Navigator is opened. Click on the tab Targets with Failed QC Procedure to see a table with the problems that appeared in the imported sequence data. You can double click on a row in the table to jump to the according position in the contig.
5 Phylogentic Analyzing with cgMLST
- Step 1: Choose from the menu Tools | Comparison Table to perform phylogenetic analysis.
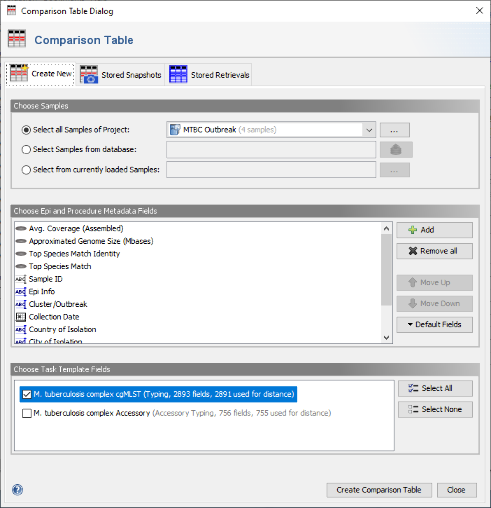
- Step 2: In the Comparison Table dialog go to the first tab "Create New". In the Choose Samples section the new previously created project (e.g., MTBC outbreak). In the Choose Genotypings Schemes section at the bottom cgMLST should already be preselected. Press the Create Comparison Table button to confirm and create the comparison table.
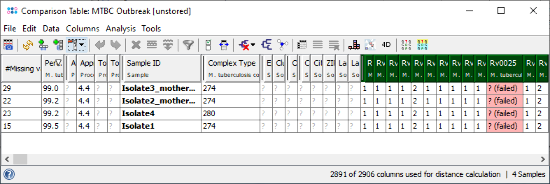
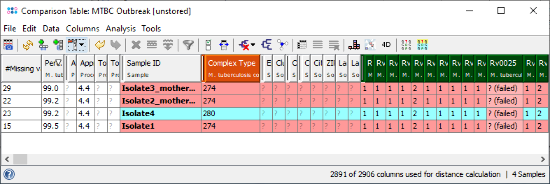
- Step 3: The comparison table is opened and shows the data for the four Samples. The columns with a green header are used for distance calculation. Some table cells contain missing values. Those appear if a cgMLST target was not found at all in an input sequence ("? (not found)"), or if the Target QC Procedure for this target has failed, e.g., because of a frame shift error ("? (failed)"). The first column in the table shows number of missing values per row.
- Step 4: Right-click on the column Complex Type (fifth column) and choose from the menu
 Set Color Groups by Column Values. Leave the upcoming dialog to defaults and confirm with OK. The Sample rows are now colored by the different cgMLST Complex Types.
Set Color Groups by Column Values. Leave the upcoming dialog to defaults and confirm with OK. The Sample rows are now colored by the different cgMLST Complex Types.
- Step 5: Press the
 Minimum Spanning Tree button in the toolbar to calculate the distances between the Samples and draw a minimum spanning tree for them. Because the table contains missing data, it must be confirmed that the missing values are ignored pairwise. Confirm with OK.
Minimum Spanning Tree button in the toolbar to calculate the distances between the Samples and draw a minimum spanning tree for them. Because the table contains missing data, it must be confirmed that the missing values are ignored pairwise. Confirm with OK.
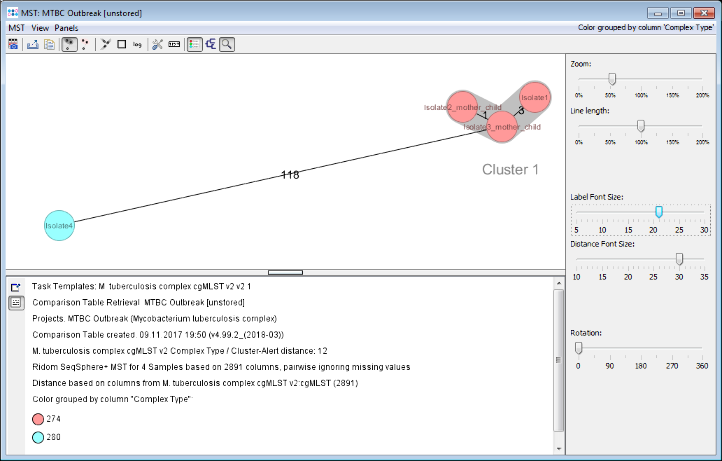
- Step 6: The minimum spanning tree is calculated for the allelic profiles of the 2,891 cgMLST targets (pairwise ignored missing values) and is shown in a new window. The controls on the right can be used to adjust the layout of the tree. By default the nodes are again colored by the Complex Type (CT) and it can be easily seen that 3 of the 4 isolates have the same Complex Type. Just Isolate4 belongs to a different Complex Type.
- Two conclusions can be drawn from this tree.
- One of the isolates without epidemiological information (Isolate1) has the same Complex Type and a close distance to two other isolates (3-4 alleles) with an epidemiological link (mother/child). This indicates epidemiological relationship.
- The other isolate without epidemiological information (Isolate4) has a different Complex Type and a distance of 118 alleles to the three other isolates. This clearly excludes recent transmission via one of those three patients.
The MST can be exported by clicking the ![]() Export MST icon of the toolbar. In the upcoming Export MST file dialog choose as file type the Scalable Vector Graphics (*.svg) or Windows Enhanced Metafile (*.emf) format. Note that EMF or SVG are vector graphics formats and therefore suited for finishing publication ready figures. EMF files can imported and scaled by MS PowerPoint. SVG files can be edited, e.g., with Adobe Illustrator or the open-source InkScape tool (once the file is loaded first ungroup all objects).
Export MST icon of the toolbar. In the upcoming Export MST file dialog choose as file type the Scalable Vector Graphics (*.svg) or Windows Enhanced Metafile (*.emf) format. Note that EMF or SVG are vector graphics formats and therefore suited for finishing publication ready figures. EMF files can imported and scaled by MS PowerPoint. SVG files can be edited, e.g., with Adobe Illustrator or the open-source InkScape tool (once the file is loaded first ungroup all objects).
For later usage of the comparison table and the MST a comparison table snaphsot can be stored by using the first button ![]() in the comparison table or the MST window, or the according function in the File menu.
in the comparison table or the MST window, or the according function in the File menu.
FOR RESEARCH USE ONLY. NOT FOR USE IN CLINICAL DIAGNOSTIC PROCEDURES.