

Contents
Reference Sequence
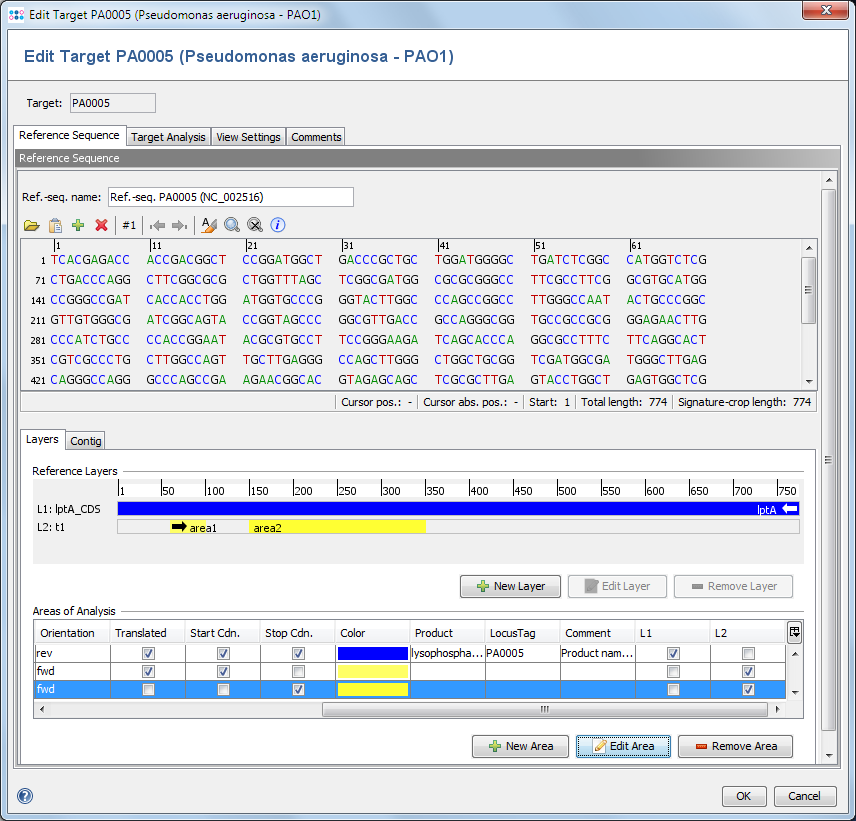
Defines a reference sequence (ref.-seq.) for the target. The main usage of a reference sequence is to orientate and crop a contig and to define variant positions. The reference sequence can be imported from a file or pasted from the clipboard.
If the Task Template was created from a reference genome, the ref.-seq. is taken from the reference core gene of this target.
Layer Settings
A layer defines a coding area that does not need to be continous. Each layer can consist of multiple areas.
ref.-seq. Areas
Areas are particularly labeled continuous regions in a reference sequence.
They can be imported from GenBank sequence file, or they can be created manually.
Areas are orientated (forward or reverse) and they can be marked as translatable.
Each area defines:
- Name
- Comment
- Type (e.g. gene, exon, non-coding)
- Product (from gb file)
- LocusTag (from gb file)
- Location
- Translatable
- Containing start codon / stop codon
- Orientation (forward/reverse)
- Area numbering offset (used for calculating the area Position)
- Color
ref.-seq. Layers
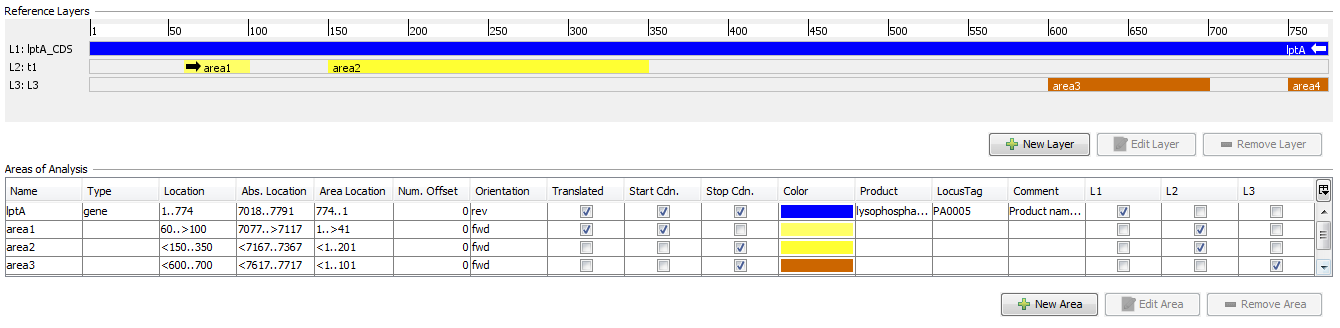
Layers are used to group one or more non-overlapping areas of a reference sequence.
By default, the first layer always covers the whole reference sequence.
Each layer defines:
- Name
- Comment
- ORF offset (+0/+1/+2)
- Orientation (forward/reverse)
- Areas belonging to this layer. All areas must have the same orientation (forward/reverse) as the layer.
Different Position Specifications
- Contig ref.-seq. Alignment
- A position of a base in the consensus is determined by the aligned position the reference sequence. If no reference sequence alignment can be done, the consensus starts with 1.
- Position
- This is the default position specification. The first base in the ref.-seq. is by default position 1. It can be redefined in the Task Template parameters.
- Absolute Position
- This position is useful when the ref.-seq. was extracted from a genome file. It gives the position of a base in the genome. If the ref.-seq. was not extracted, the absolute position is the same as the standard position.
- Area Position
- Gives the position of a base in an Area. It is calculated from the area number offset that is defined in the area and counted in the orientation of the area.
Contig Settings
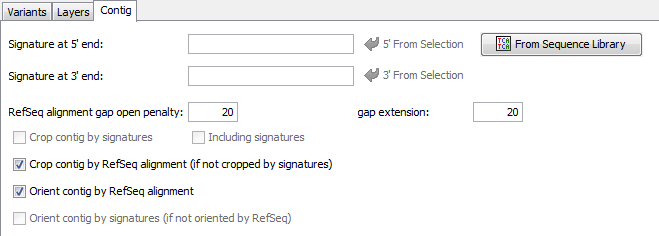
Contig Signatures
Defines the 5' and the 3' signature for a contig target. For convenience the signatures can be copied directly from a selection made in the reference sequence (buttons 5' and 3').
In addition the signatures can be imported from a multiple alignment file by using the From Sequence Library button. Again an inclusive consensus will be called and the beginning and the end of the consensus can be used as signatures. The Including signatures check-box can be used to define, if the signatures itself should belong to the contig, or not.
The signatures may contain ambiguities. In this case, the signatures will match to every base that is expressed by this ambiguity, and to the ambiguity itself. If the signature should not match the ambiguity but only to the different bases, the base characters must be grouped with [ ]. Example: W matches to A,T or W; but [AT] matches only to A or T.
ref.-seq. Alignment Settings
The gap opening penalty, and the gap extension penalty for the alignment between the consensus and the ref.-seq. can be set.
Contig Cropping and Orientation
The signatures and the reference sequence can be used to orientate and to trim the contig automatically.