

Introduction
NCBI AMRFinderPlus (Antimicrobial Resistance Gene Finder Plus; citation) is used to find AMR-specific genes and proteins from the NCBI Bacterial Antimicrobial Resistance Reference Gene Database (BioProject PRJNA313047). Furthermore , genes related to biocide and stress resistance, general efflux, virulence, or antigenicity are searched ('plus' option). In SeqSphere+, only the BLASTX protein search from translated nucleotide sequences of the assembly contigs against the AMR protein database is used to identify AMR proteins (the HMMER search to detect new resistances is currently not used in SeqSphere+).
NCBI AMRFinderPlus is deployed with the SeqSphere+ installation, but requires that the SeqSphere+ client is running on Linux or on Windows 10 with installed Windows Subsystem for Linux (WSL).
The predefined task template 'NCBI AMRFinderPlus' can be downloaded with Linux or WIN 10 clients from the Task Template Sphere for all organisms except Mycobacterium. For Escherichia coli the specific task template 'E. coli NCBI AMRFinderPlus' is listed for download in the Task Template Sphere.
Alternatively, AMRFinderPlus can also be called with those clients as standalone function in the tools menu. Once downloaded the task template is stored on the server. Therefore, this task template now becomes also available for clients without installed WSL. If such a client tries to execute a pipeline that makes use of a ‘NCBI AMRFinderPlus’ task template an error will be elicited (also when the Test Pipeline Script function is performed). However, viewing AMRFinderPlus results produced with an appropriate client is possible with all clients.
Disclaimer: Users of AMRFinderPlus or its supporting data files are cautioned that presence of a gene encoding an antimicrobial resistance (AMR) protein or resistance causing mutation does not necessarily indicate that the isolate carrying the gene is resistant to the corresponding antibiotic. AMR genes must be expressed to confer resistance. Many AMR proteins reduce antibiotic susceptibility somewhat, but not sufficiently to cross clinical breakpoints. Meanwhile, an isolate may gain or lose resistance to an antibiotic by mutational processes, such as the loss of a porin required to allow the antibiotic into the cell. For some families of AMR proteins, especially those borne by plasmids, correlations of genotype to phenotype are much more easily deciphered, but users are cautioned against over-interpretation (cited from AMRFinderPlus documentation).
Task Entry Overview
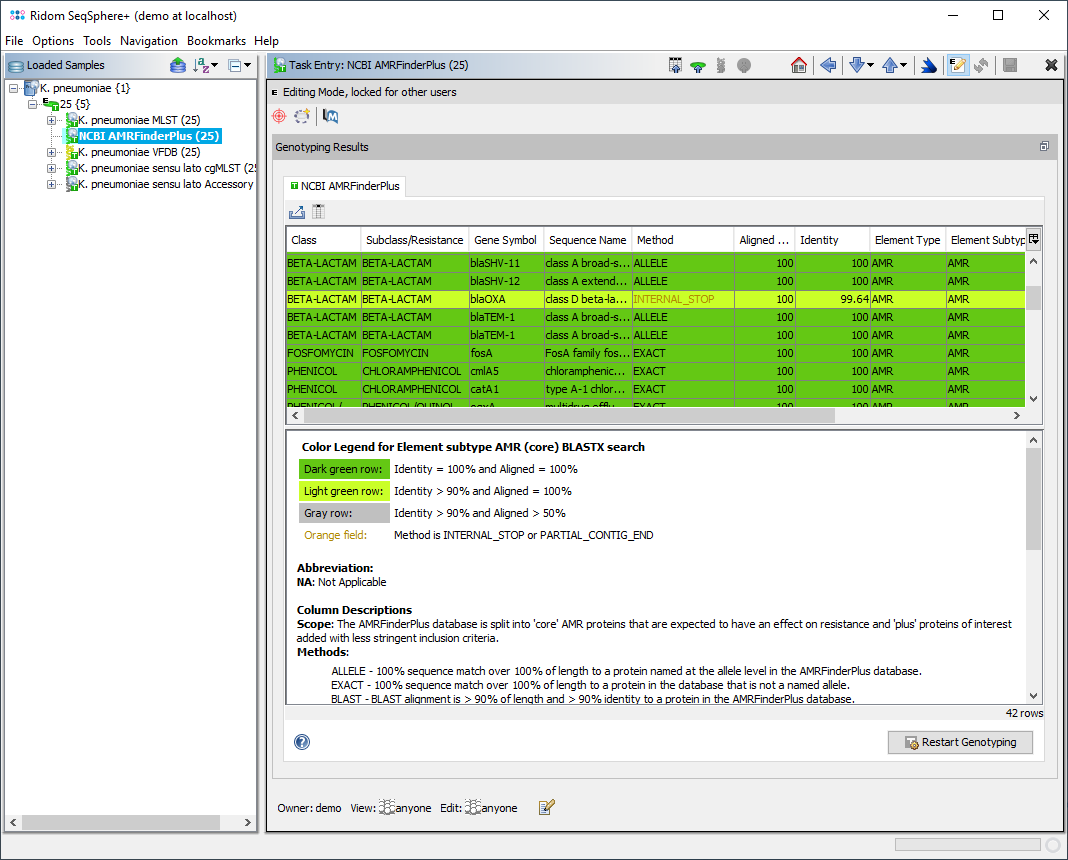
When a NCBI AMRFinderPlus task entry is processed, SeqSphere+ starts the deployed AMRFinderPlus. The Task Entry Overview of the processed task entry shows a table with the AMRFinderPlus output with the target genes found. If multiple matches for a target are found on different locations, each match is listed as separate row in the table.
The table rows for Element subtype AMR (core) and for E. coli Class STX are colored by the percental identity and alignment overlap with allele in database using the following thresholds:
- Dark green row: Identity = 100% and Aligned Overlap = 100%
- Light green row: Identity ≥ 90% and Aligned Overlap = 100%
- Gray row: Identity ≥ 90% and Aligned Overlap ≥ 50%
If the Method column contains INTERNAL_STOP or PARTIAL_CONTIG_END the table cells are highlighted in orange to indicate a warning.
The table contains the following columns:
- Class - For AMR genes this is the class of drugs that this gene is known to contribute to resistance of.
- Subclass - If more specificity about drugs within the drug class is known it is elaborated here.
- Gene symbol - Gene or gene-family symbol for nucleotide hit. For point mutations it is a combination of the gene symbol and the SNP definition separated by "_"
- Sequence name - Full-text name for the nucleotide.
- Method - Type of hit found by AMRFinder.
- ALLELE - 100% sequence match over 100% of length to a protein named at the allele level in the AMRFinderPlus database.
- EXACT - 100% sequence match over 100% of length to a protein in the database that is not a named allele.
- BLAST - BLAST alignment is > 90% of length and > 90% identity to a protein in the AMRFinderPlus database.
- PARTIAL - BLAST alignment is > 50% of length, but < 90% of length and > 90% identity to the reference, and does not end at a contig boundary.
- PARTIAL_CONTIG_END - BLAST alignment is > 50% of length, but < 90% of length and > 90% identity to the reference, and the break occurs at a contig boundary indicating that this gene is more likely to have been split by an assembly issue.
- INTERNAL_STOP - Translated blast reveals a stop codon that occurred before the end of the protein.
- POINT - Point mutation identified by blast.
- % Coverage of reference sequence - % of reference covered by blast hit.
- % Identity to reference sequence - % nucleotide identity for nucleotide reference.
- Element type - AMRFinder+ genes are placed into functional categories based on predominant function AMR, STRESS, or VIRULENCE.
- Element subtype - Further elaboration of functional category into (ANTIGEN, BIOCIDE, HEAT, METAL, PORIN) if more specific category is available, otherwise the element is repeated.
- Scope - The AMRFinderPlus database is split into 'core' AMR proteins that are expected to have an effect on resistance and 'plus' proteins of interest added with less stringent inclusion criteria. These may or may not be expected to have an effect on phenotype.
- Core: this subset includes highly curated, AMR-specific genes and proteins from the Bacterial Antimicrobial Resistance Reference Gene Database (BioProject PRJNA313047), plus point mutations. The sources of input for this curated database include: 1) allele assignments, 2) exchanges with other external curated resources, 3) reports of novel antimicrobial resistance proteins in the literature.
- Plus: this subset includes genes related to biocide and stress resistance, general efflux, virulence, or antigenicity. These genes are only shown if the --plus option is used.
More details can be found int the NCBI AMRFinderPlus documentation.
Below the table a colored threshold legend, version information, and citation(s) are stated.
Result Fields
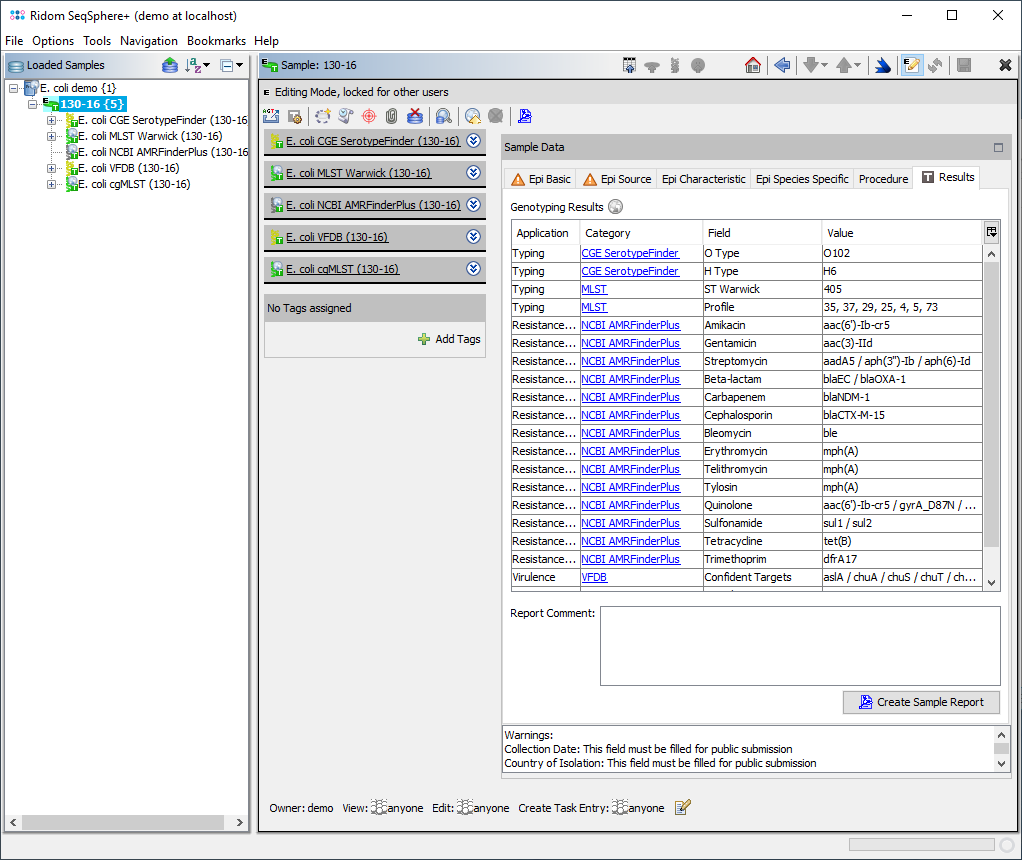
The stored result fields are per following AMR (core) subclass aggregated and therefore may contain more than one target that are then delimited by '/' (e.g., "Beta-lactam" = "blaSHV-11 / blaSHV-12 (ESBL) / blaTEM-1"):
| Class | Subclass/Resistance |
|---|---|
| AMINOGLYCOSIDE | AMIKACIN |
| AMINOGLYCOSIDE | GENTAMICIN |
| AMINOGLYCOSIDE | HYGROMYCIN |
| AMINOGLYCOSIDE | KANAMYCIN |
| AMINOGLYCOSIDE | KASUGAMYCIN |
| AMINOGLYCOSIDE | SPECTINOMYCIN |
| AMINOGLYCOSIDE | STREPTOMYCIN |
| AMINOGLYCOSIDE | TOBRAMYCIN |
| AVILAMYCIN | AVILAMYCIN |
| BACITRACIN | BACITRACIN |
| BETA-LACTAM | AMPICILLIN |
| BETA-LACTAM | BETA-LACTAM |
| BETA-LACTAM | CARBAPENEM |
| BETA-LACTAM | CEPHALOSPORIN |
| BETA-LACTAM | CEPHALOTHIN |
| BETA-LACTAM | METHICILLIN |
| BLEOMYCIN | BLEOMYCIN |
| BLEOMYCIN | ZORBAMYCIN |
| COLISTIN | COLISTIN |
| FLUOROQUINOLONE | FLUOROQUINOLONE |
| FOSFOMYCIN | FOSFOMYCIN |
| FUSIDIC ACID | FUSIDIC ACID |
| GLYCOPEPTIDE | VANCOMYCIN |
| LINCOSAMIDE | LINCOSAMIDE |
| MACROLIDE | ERYTHROMYCIN |
| MACROLIDE | TELITHROMYCIN |
| MACROLIDE | TYLOSIN |
| MUPIROCIN | MUPIROCIN |
| NITROIMIDAZOLE | NITROIMIDAZOLE |
| PHENICOL | CHLORAMPHENICOL |
| PHENICOL | FLORFENICOL |
| PHENICOL | PHENICOL |
| OXAZOLIDINONE | OXAZOLIDINONE |
| PLEUROMUTILIN | PLEUROMUTILIN |
| PLEUROMUTILIN | TIAMULIN |
| QUINOLONE | QUINOLONE |
| RIFAMYCIN | RIFAMPIN |
| RIFAMYCIN | RIFAMYCIN |
| STREPTOGRAMIN | STREPTOGRAMIN |
| STREPTOTHRICIN | STREPTOTHRICIN |
| SULFONAMIDE | SULFONAMIDE |
| TETRACENOMYCIN | TETRACENOMYCIN |
| TETRACYCLINE | TETRACYCLINE |
| THIOSTREPTON | THIOSTREPTON |
| TRIMETHOPRIM | TRIMETHOPRIM |
| TUBERACTINOMYCIN | VIOMYCIN |
Those fields are filled-in by the AMRFinderPlus output column 'Subclass/Resistance' of all confident calls, i.e., green rows. 'Classes' that appear as result in 'Subclass/Resistance' column (like AMINOGLYCOSIDE) are resolved into the specific subclasses. If the result for Subclass BETA-LACTAM contains the word extended in the column Sequence Name, then the postifx (ESBL) is appended to the result field value. If multiple matches for a target are found on different locations, the targets matched are concatenated with "," (e.g., "Beta-lactam" = "blaSHV-11 / blaSHV-12 (ESBL) / blaTEM-1, blaTEM-1").
Additionally to the fields listed above, the Escherichia coli specific task template 'E. coli NCBI AMRFinderPlus' defines the two result fields STX1 and STX1.
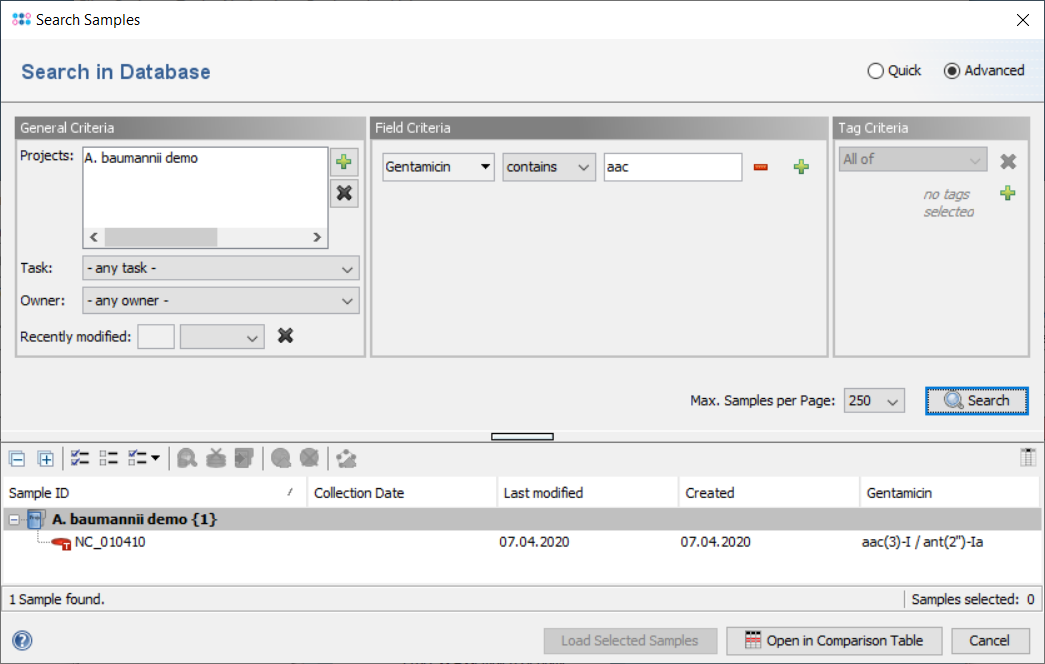
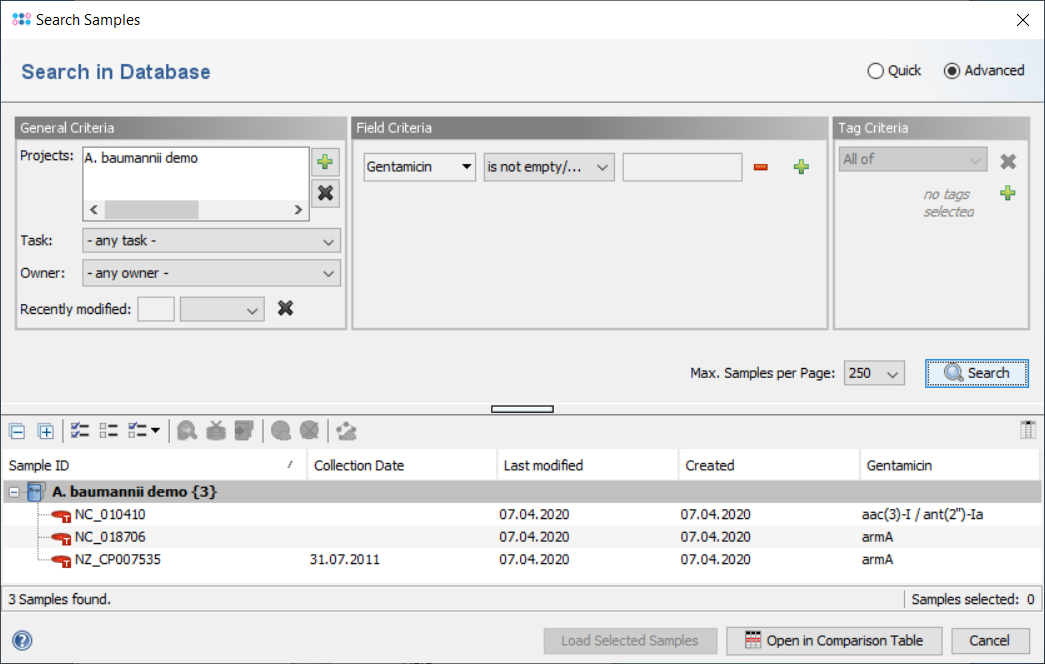
The result fields are shown in the result tab of the Sample Overview. By clicking the NCBI AMRFinderPlus category links all details for this sample can be viewed. The result fields can be selected from the NCBI AMRFinderPlus entry for searching under 'Field Criteria' in the advanced mode of the sample search dialog. The search can be done either by using the operator 'contains' for specific targets or by using the operator 'is not empty' for any target in this field.
The result fields can also be retrieved for a Comparison Table and for exporting metadata. If the NCBI AMRFinderPlus Task Template is chosen in the Create Comparison Table dialog, then the results fields are not used for distance calculation but are shown with gray column headings for descriptive purpose only. For a better overview it is recommended to use the command Columns | Remove Resistance/Virulence Genotyping Columns where All Values Are Missing to get rid of those columns that are for all samples empty.
If a resistance profile (presence/absence) comparison of several samples is intended to be done, then the command Columns | Transform Resistance/Virulence Genotyping Columns to Absence/Presence (+/-) may be used. Alternatively handle missing values as an own category when building trees. Next the command Columns | Select Genotyping Schemes for Distance Calculation must be elicited and in the upcoming dialog AMRFinderPlus must be selected and all other schemes should best be deselected from distance calculation. If data were not transformed, then once the command for calculating a tree was elicited in the upcoming missing values dialogue the option Missing Values are Own Category must be selected.
Tools Menu Function
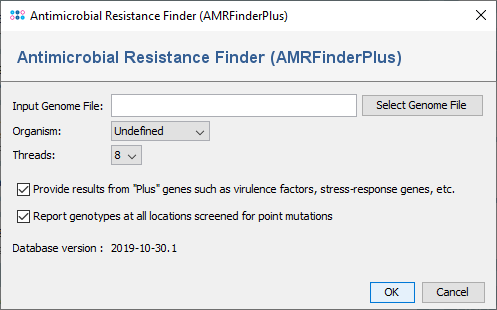
The NCBI AMRFinderPlus can also be invoked manually for a FASTA file using the menu function Tools | Genome Utilities | Antimicrobial Resistance Finder (AMRFinderPlus). The following options in the dialog can be set:
- Organism: This field is selected as Undefined by default and it contains values of supported organisms. Selecting organism enables screening for point mutations which suppresses the reporting of some that are extremely common in selected organism.
- Plus: AMRFinderPlus splits the database into two subsets and the one that includes genes related to biocide and stress resistance, general efflux, virulence, or antigenicity will be only shown if this option is selected.
- Point Mutations: For supported organisms, they are identified by BLAST alignments that cover at least 50% of the reference at 90% identity. Offsets are calculated relative to the beginning of the reference and reported in that coordinate system. That is if there are indels within the query sequence the coordinates of the point mutation will reflect the offset from the start codon in the reference rather than in the query sequence.
Runtimes
The following table contains the measured NCBI AMRFinderPlus runtimes on a quad core (8 threads) desktop with 32GB RAM (using Windows Subsystem for Linux). Two different thread settings were used: single-threaded and parallel using all cores/hyperthreads. The speedup when using multiple threads depends on the number of (large) contigs as the BLASTX search is parallelizable per contig. Therefore, finished genomes consisting of a single contig take longer than draft genomes.
| Strain | Contig Count | Genome Size (Mbases) | Runtime with 1 thread | Runtime with 8 threads |
|---|---|---|---|---|
| Staphylococcus aureus COL (draft genome) | 36 | 2.8 | 122s | 43s |
| Escherichia coli Sakai (NCBI) | 1 | 5.5 | 286s | 287s |
| Escherichia coli Sakai (draft genome) | 248 | 5.3 | 291s | 87s |
| Pseudomonas aeruginosa PAO1 (draft genome) | 165 | 6.2 | 423s | 107s |