

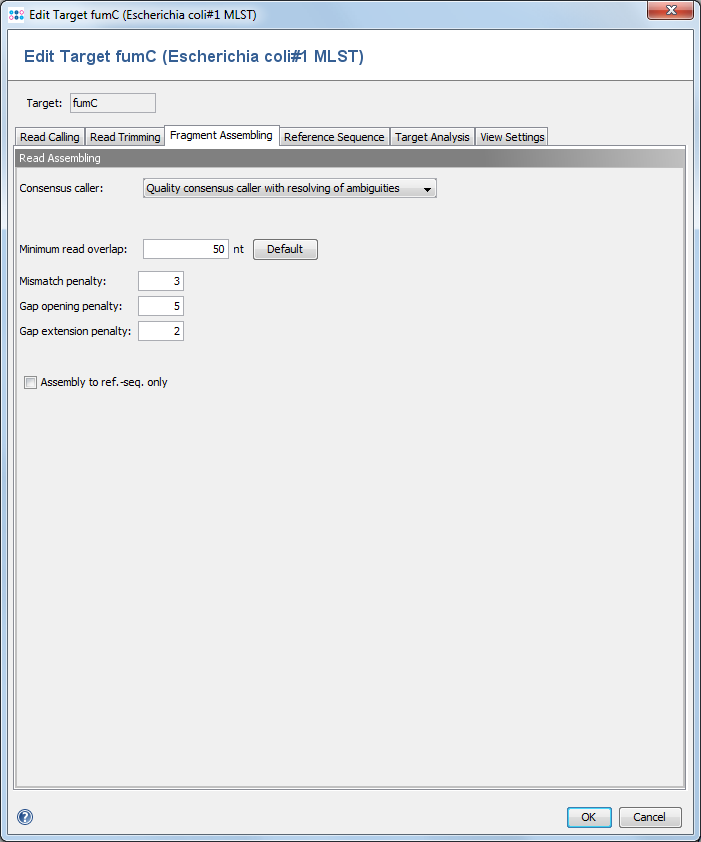
This dialogs defines the consensus calling and assembling functions. ![]() This feature is only available for Sanger sequencing data.
This feature is only available for Sanger sequencing data.
Contents
Consensus caller
The consensus caller for the contigs of an assembly can be chosen here. Currently there are five different consensus callers available.
Quality Consensus Caller Without Resolving of Ambiguities
This algorithm calculates the consensus base for a sequence column with Bayes formula. The quality of the read bases and the read orientation are used in this calculation. In a first step separate quality values for forward and reverse directions are calculated with Bayes formula. In a second step Bayes formula is used again to calculate a combined consensus quality value. Ambiguity bases are counted as separate base types and are not resolved. Each gap is given the quality of 20, and if the quality sum of the gaps exceeds the quality sum of all base calls, the consensus will have a gap at this position.
This consensus calling method should be used for input sequences with possible heterozygous positions (determined by a heterozygot caller) and with quality values related to an error probability by the formula <math> quality = -10 * \log_{10} (error-probability) </math>
Quality Consensus Caller With Resolving of Ambiguities
This algorithm is very similar to the first one (Quality Consensus Caller Without Resolving of Ambiguities). The only difference is the treatment of ambiguities: ambiguous bases are resolved here, for example a S counts as C and G.
This consensus calling method should be used for input sequences (usually with a heterozygot caller turned off) with quality values related to an error probability by the formula <math> quality = -10 * \log_{10} (error-probability) </math>
Majority Consensus Caller
This algorithm gives each base the same weight. The consensus base is determined by the majority base type of a column. Ambiguities cast a vote for each possible base call they represent, for example an S counts for C and G. If there is no unique majority base, an ambiguity including all bases will be called. If there are 66% or more gaps at one column position, the consensus will have a gap at this position.
This consensus calling method should be used for input sequences without quality values
Strict Consensus Caller
This algorithm performs a consensus calling allowing only a very limited number of mismatches. In general the algorithm performs a majority consensus call. However, all consensus columns with a coverage of only one read get a N consensus base. The table below shows the maximum allowed number of mismatches in a column for a given coverage. More mismatches will lead to a N call. All ambiguous base calls are treated as N. If there are 66% or more gaps at one column position, the consensus will have a gap at this position.
This consensus calling method should be used for forensic DNA typing
Inclusive Consensus Caller
This algorithm includes every base type in a column. The consensus base is the smallest ambiguity base covering all read bases. If there are 66% or more gaps at one column position, the consensus will have a gap at this position.
Minimum read overlap
This parameter can be used to configure the minimum overlap that two reads must share, before they are aligned together. Under normal circumstances the default value of 50 works fine.
Mismatch / Gap Opening / Gap Extension Penalties
The penalty values for the assembling algorithm. The defaults are 3 / 5 / 2.
Assemble to ref.-seq. only
Instead of aligning all reads to each other in an assembling process, the reference sequence only can be used for guiding the building of an assembly (this will speed up the process).