

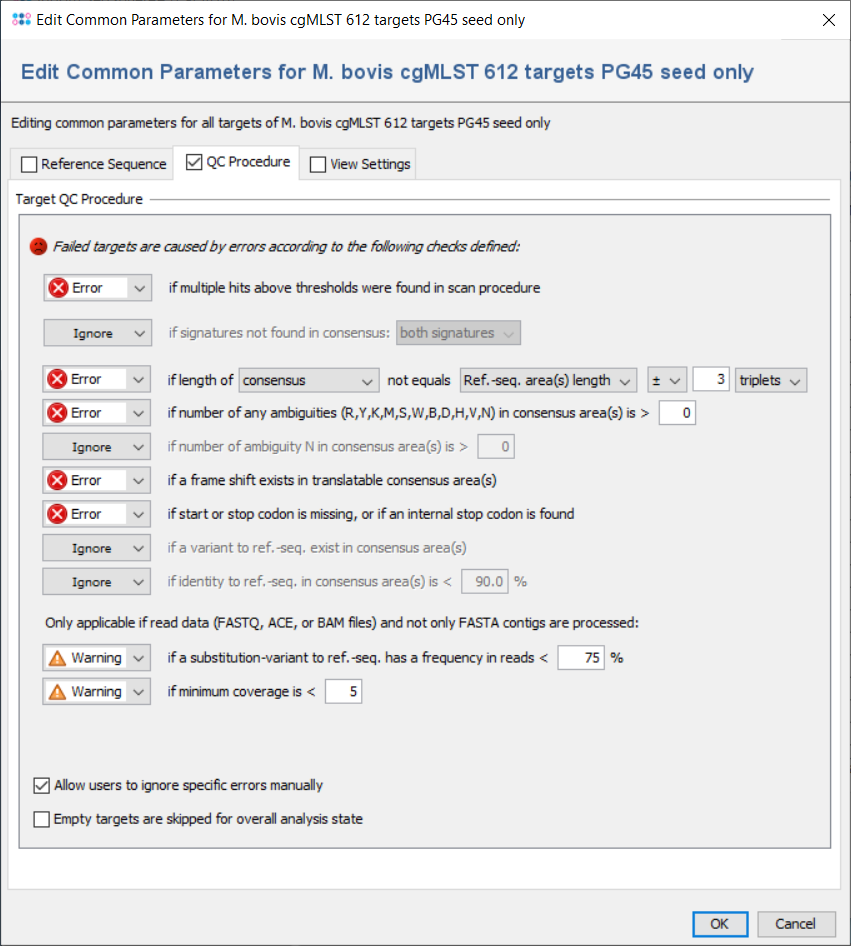
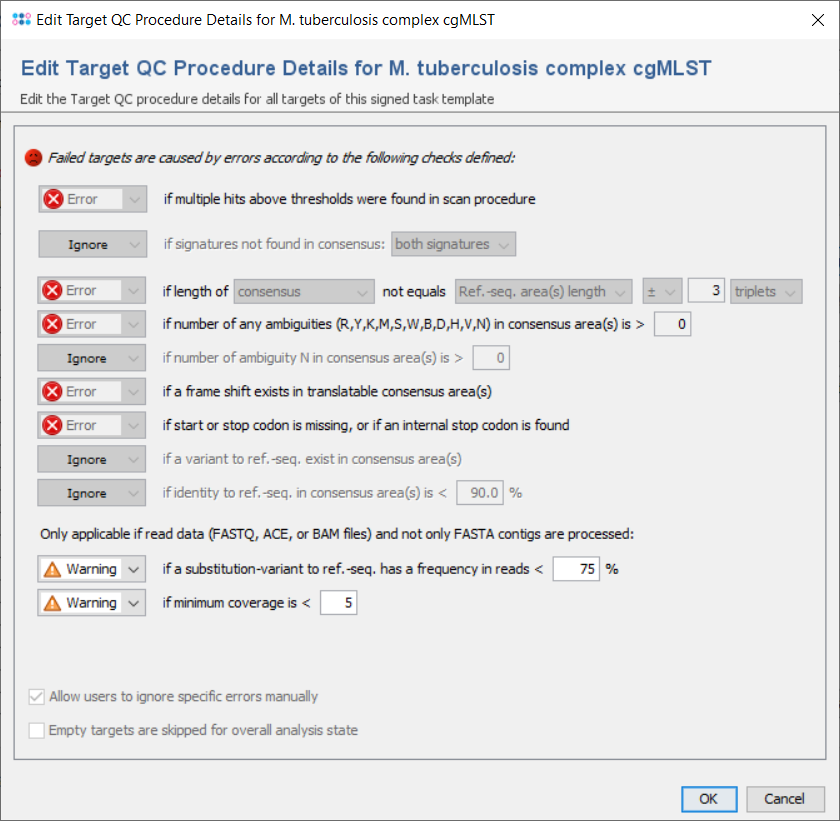
All targets are automatically checked for the quality issues that were defined in the Analysis Parameters of the Task Template. Each check can set to
- Ignore:
- Check is not done at all.
- Warning:
- If target fails the check it is rated as
 good with warnings.
good with warnings.
- If target fails the check it is rated as
- Error:
- If target fails the check it is rated as
 failed.
failed.
- If target fails the check it is rated as
But default, the parameters are the same for all targets, but they can also be defined targets-specific. If a target has multiple sequences and if this is allowed by the first check), all further checks are performed for the first one only,
The checks always work on the areas of the layers that are defined for the target in the task template. Usually (e.g., in cgMLST), a target has only one layer with a single area that covers the whole target sequence, and defines its orientation (forward/reverse). Therefore, the terms 'layer' and 'consensus area(s)' are in most cases equivalent to the target sequence.
The following checks can be set to Ignore/Warning/Error:
- if multiple hits above thresholds were found in scan procedure
- Defines if multiple hits above the identity and aligned thresholds are allowed and imported in the target scan procedure, or not.
- if signatures not found in consensus
- Defines that the 3` signature, the 5`signature, or both must be found in the consensus.
- if length of consensus not equals ref.-seq. length with a specified tolerance
- Defines that the trimmed consensus, or all bases of the consensus that are covered by a ref.-seq. area, must have a specific length, or the same length as the reference sequence in all areas of this layer. A tolerance range for the length comparison can be set in bases or codons.
- if minimum mean base quality is above a specified value
- Defines the allowed minimum mean base quality, calculated for all bases in the consensus areas of this layer.
 This feature is only available for Sanger sequencing data.
This feature is only available for Sanger sequencing data.
- if number of low quality bases is above a specified value
- Defines the allowed maximum number of low quality bases in the consensus areas of this layer. The threshold for a low quality can be specified. This feature is only available for Sanger sequencing data.
- if number of any ambiguities (R,Y,K,M,S,W,B,D,H,V,N) is above a specified value
- Defines the allowed maximum number of any ambiguities in the consensus areas of this layer
- if number of any ambiguity N is above a specified value
- Defines the allowed maximum number of the ambiguity N in the consensus areas of this layer
- if a frame shift exists in translatable consensus area(s)
- Defines that the number of insertion(s) in comparison to ref. seq. minus the number of deletion(s) must be dividable by 3.
- if start or stop codon is missing, or if an internal stop codon is found
- Defines that the translation of the consensus areas according to the layer must contain a start codon and a stop codon only at the expected positions, if they are defined in the areas of an translatable layer. Therefore, if layer and target sequence are identical as it is usually the case this check fails if no start codon is found at the begin, no stop codon is found at the end, or a stop codon is found at a wrong position in a target.
- if a variant to ref.-seq. exist in consensus area(s
- Demand that no differences exist between the consensus and the ref.-seq. in the areas of this layer.
- if identity to ref.-seq. is below a specified value
- Identity is calculated as the rational difference between consensus and reference sequence in an alignment. Differences are gaps and mismatches. Ambiguities never match to bases, but to the same ambiguity
- if a substitution-variant to ref.-seq. has a frequency in reads is below a specified value
- Defines that substitution-variants to the ref.-seq. must be confirmed by a specific percentage of reads that cover this position (Default: Warning if below 75%).
- if minimum coverage is below a specified value (Whole Genome Sequencing)
- Defines the coverage that is demanded for every base (is not average coverage) in the consensus areas of this layer in any reading direction (Default: warning if below 5). This feature is only available for whole genome sequencing data.
- if minimum coverage is below a specified value (Sanger Sequencing)
- Defines the coverage that is demanded for the bases in the consensus areas of this layer, and optionally if both reading directions (forward and reverse) should be covered in the reads. The coverage is defined by percentage for to the base count in the consensus areas. To handle badly covered ends of the contig, it can be defined that the uncovered positions (if demanded coverage <100%) may only appear on the ends of the contig. This feature is only available for Sanger sequencing data.
Processing Options
- Allow users to ignore specific analysis problems
- If this option is checked, an Ignore button is added to the analysis problem table in sequence view.
- Empty targets are skipped for overall analysis state
- By default, targets without sequence will set the analysis state of the whole task entry to 'bad'. If this option is set, any targets without sequence will be ignored during analysis and will have no effect on the analysis state of the task entry.