

Contents
Introduction
The Chromosome & Plasmids Overview task can be used to reconstruct and type plasmid sequences in the assembled contigs and to combine them with the results of the AMRFinderPlus, MobileElementFinder, and the VFDB tasks. For reconstruction and typing of the plasmids the MOB-suite is used. Reconstructed plasmids are compared against the MOB-suite database that consists of more than 17.750 NCBI reference plasmids.
![]() Important:
Important:
- If SeqSphere+ is installed on Windows, MOB-suite requires the Windows Subsystem for Linux (WSL).
- If SeqSphere+ is installed on Linux, MOB-suite must once be installed by calling the installation of Bioinformatic Tools on Linux.
- The AMRFinderPlus, MobileElementFinder, and VFDB task templates must be added to the project to provide the antimicrobial resistance (AMR) targets, integrative mobile genetic elements (iMGE), and virulence factor (VF) targets for the Chromosome & Plasmids Overview task.
Plasmids are mobile genetic elements (MGEs), which allow for rapid evolution and adaption of bacteria to new niches through horizontal transmission of novel traits to different genetic backgrounds. The MOB-suite is designed to be a modular set of tools for the typing and reconstruction of plasmid sequences from WGS assemblies.
SeqSphere+ uses two tools from the MOB-suite:
- MOB-recon
- Reconstructs individual plasmid sequences from draft genome assemblies using the clustered plasmid reference databases provided by MOB-cluster. In SeqSphere+ not used for circular contigs (topology=circular in FASTA header). Circular contigs with a size below 500kb are regarded as plasmids and analyzed with the MOB-typer module only. MOB-typing is automatically generated for all plasmids reconstructed by MOB-recon.
- MOB-typer
- Provides in silico predictions of the replicon family, relaxase type, Mash Neighbor Distance and NCBI Accession, predicted host range and associated PMID(s), and predicted transferability of the plasmid. Also assigns for a plasmid Primary and Secondary Cluster IDs. Plasmids are grouped into clusters using complete-linkage clustering (0.06 for primary and 0.025 for secondary cluster ID Mash distance) and the cluster code accessions provided by the tool provide an approximation of operational taxonomic units (OTUs). Usually, due to incompatibility an isolate can not harbor plasmids with the same primary cluster ID. The same secondary ID in the plasmids of two different isolates is indicative of a transmission event. Both thresholds are optimized for short read data. If reconstructed plasmids exceed the Mash distance for primary cluster assignment, then they will be assigned a primary cluster ID in the format novel_{md5} where the md5 hash is calculated based on the sequences belonging to that reconstructed plasmid. Only absolute identical sequences would get the same novel_{md5} name.
SeqSphere checks the assembled contigs. Contigs that are longer than a threshold of 500.000 bases are assumed to be chromosome sequences and are therefore excluded from MOB-recon and -typer processing.
If a contig that is shorter than the threshold contains the term topology=circular in the name it is assumed to be a closed plasmid (e.g. PacBio data). Those contigs are directly passed to MOB-typer. All other contigs that are shorter than the threshold above, are processed with MOB-recon to reconstruct plasmid sequences.
Finally, the results are combined with the reliable ("green") AMRFinderPlus, CGE MobileElementFinder, and VFDB results (if those exist for the sample), to show the resistance, iMGE, and virulence markers on the chromosome & plasmid level.
Advantages and Disadvantages of Rep- and Mob-Typing and Primary & Secondary Cluster IDs
Replicon typing
- targets genetic elements of the replicon region (encoding replication machinery)
- plasmids with similar replication machinery are often unstable to stably co-exist within the same host cell (traditionally used to classify plasmids into incompatibility [Inc] groups)
- individual plasmids can contain multiple replicons
- not all plasmids contain a (known) replicon region (e.g., in Enterobacteriaceae about 75% and Firmicutes about 50%)
Mob typing
- exploits the conserved N-terminal sequence of the relaxase proteins encoded by transmissible plasmids
- types only transmissible plasmids (e.g., about 50% of gamma-Proteobacterial and about 35% of Firmicutes plasmids)
- individual transmissible plasmids usually harbor just one relaxase
- compared with replicon typing, MOB typing classifies plasmids at lower resolution
Primary and Secondary Cluster ID typing
- Primary and Secondary Cluster IDs are based on MASH distances. MASH dist allows for plasmids with considerable differences in size to be grouped together
- a clustering threshold of 0.06 for primary cluster designation (Primary Cluster ID) for plasmid reconstruction, host-range prediction and broader surveillance, along with a nested secondary cluster designation (Secondary Cluster ID) at 0.025 to recognize near duplicate plasmids is used. The 0.025 threshold was selected for near duplicates since any lower threshold would result in draft plasmids potentially assigned to a different cluster than a complete version of the plasmid
- the ID has per se no biological meaning
- is applicable to all plasmids, has a higher resolution than rep- and MOB-typing and is more representative for the whole plasmid
Combining AMR, iMGE, and VF with Chromsomes/Plasmids
NCBI AMRFinderPlus
For the found chromosome(s) and plasmid(s), the contig IDs are compared with the NCBI AMRFinderPlus results if available for the sample. If the chromosome or plasmids include a contig that contains a reliable ("green") AMR target, the target is listed in the AMRFinderPlus Targets column. If it contains a priority AMR target, the target is listed in the AMRFinderPlus Priority Targets column and highlighted in red.
A target is considered as priority AMR target, if it belongs to one of these categories:
- subclass CARBAPENEM
- postfix (ESBL), i.e. subclass BETA-LACTAM with "extended" in the name
- postfix (AmpC), i.e. subclass BETA-LACTAM with "class C" in nthe ame
- subclass COLISTIN
- subclass VANCOMYCIN
- subclass METHICILLIN
The combined results are stored in the result fields Plasmid AMRFinderPlus Targets, Plasmid AMRFinderPlus Priority Targets, Chromosome AMRFinderPlus Targets, and Chromosome AMRFinderPlus Priority Targets.
CGE MobileElementFinder
For the found chromosome and plasmid(s) that contain reliable priority AMR targets, it is also checked if they correlate with found reliable ("green") iMGEs if such results are available for the sample. If those integrative MGE(s) contain priority AMR target(s) an identifier for the combination of iMGE name(s) and AMR target(s) is listed in the column iMGE(s) Priority AMR. The corresponding iMGE accessions are listed in the column iMGE(s) Priority AMR Accessions.
The format of the combined identifier reflects the different scenarios:
- Multiple Priority AMR Targets can be part of one iMGE
- Example cn_34764_IS5075_blaCTC-M-27_blaNDM-1
- Priority AMR Targets of one plasmid/chromosome can be part of different iMGEs
- Example: cn_1217_IS26_blaNDM-1, cn_34764_IS5075_blaCTX-M-27
- The same Priority AMR Target can be part of multiple iMGEs (due to frequent nested nature of iMGEs)
- Example: cn_34764_IS5075_blaCTX-M-27 (cn_10660_IS26_blaCTX-M-27)
The identical Priority AMR Target and iMGE combination might be carried multiple times on a plasmid/chromosome (amplification; e.g. 21222 blaCTX-M-27 plasmid)
- Example: cn_1217_IS26_blaNDM-1, cn_1217_IS26_blaNDM-1, cn_1217_IS26_blaNDM-1
The combined results are stored in the result fields Plasmid iMGE(s) Priority AMRs and Chromosome iMGE(s) Priority AMRs.
VFDB
For the found chromosome and plasmid(s), the contig IDs are compared with the reliable ("green") VFDB results if available for the sample. If the chromosome or plasmid(s) include a contig that contains a VFDB target, the virulence factor (VF) is listed in the VFDB Targets column.
The combined results are stored in the result fields Plasmid VFDB Targets and Chromosome VFDB Targets.
Task Entry Overview
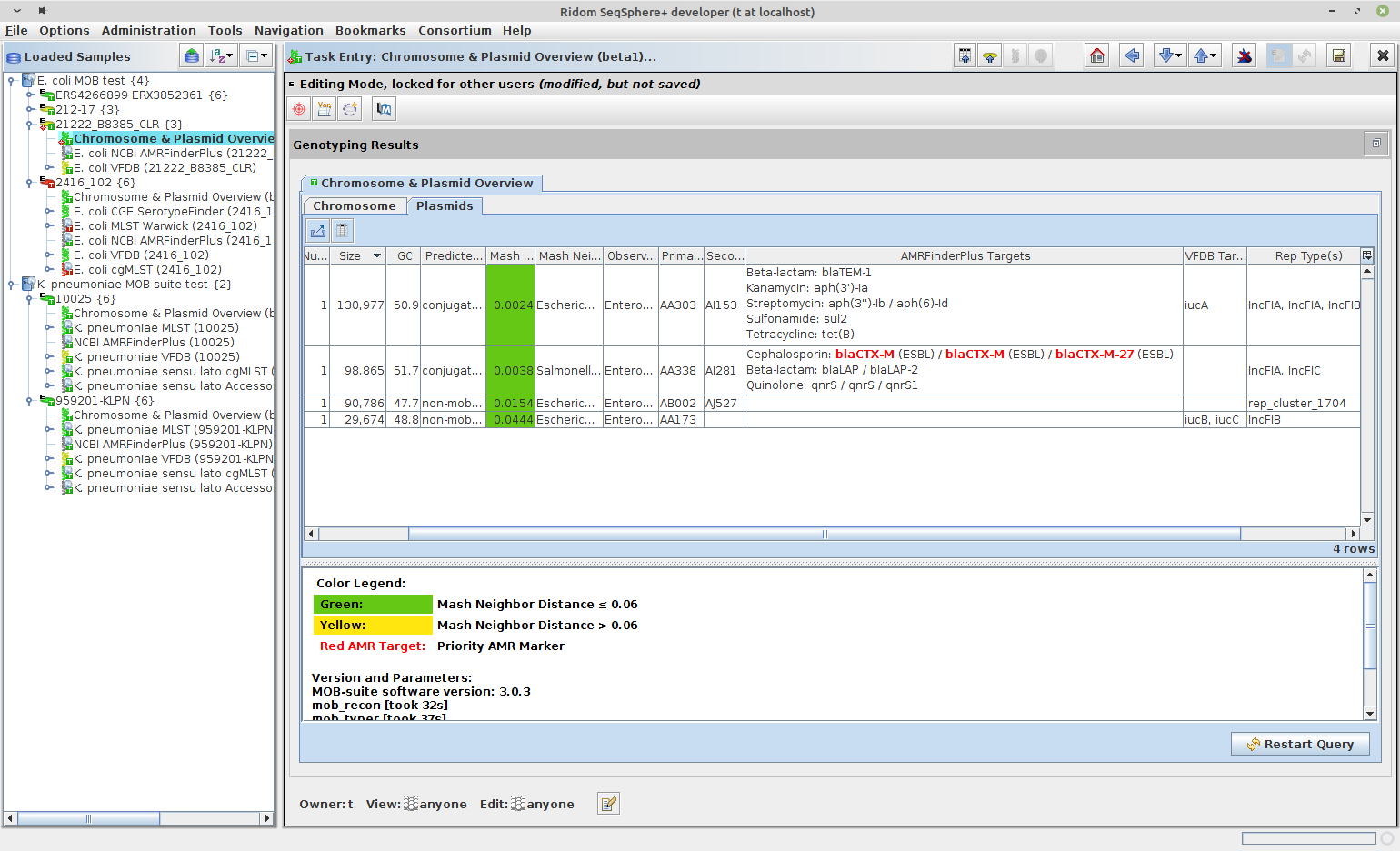
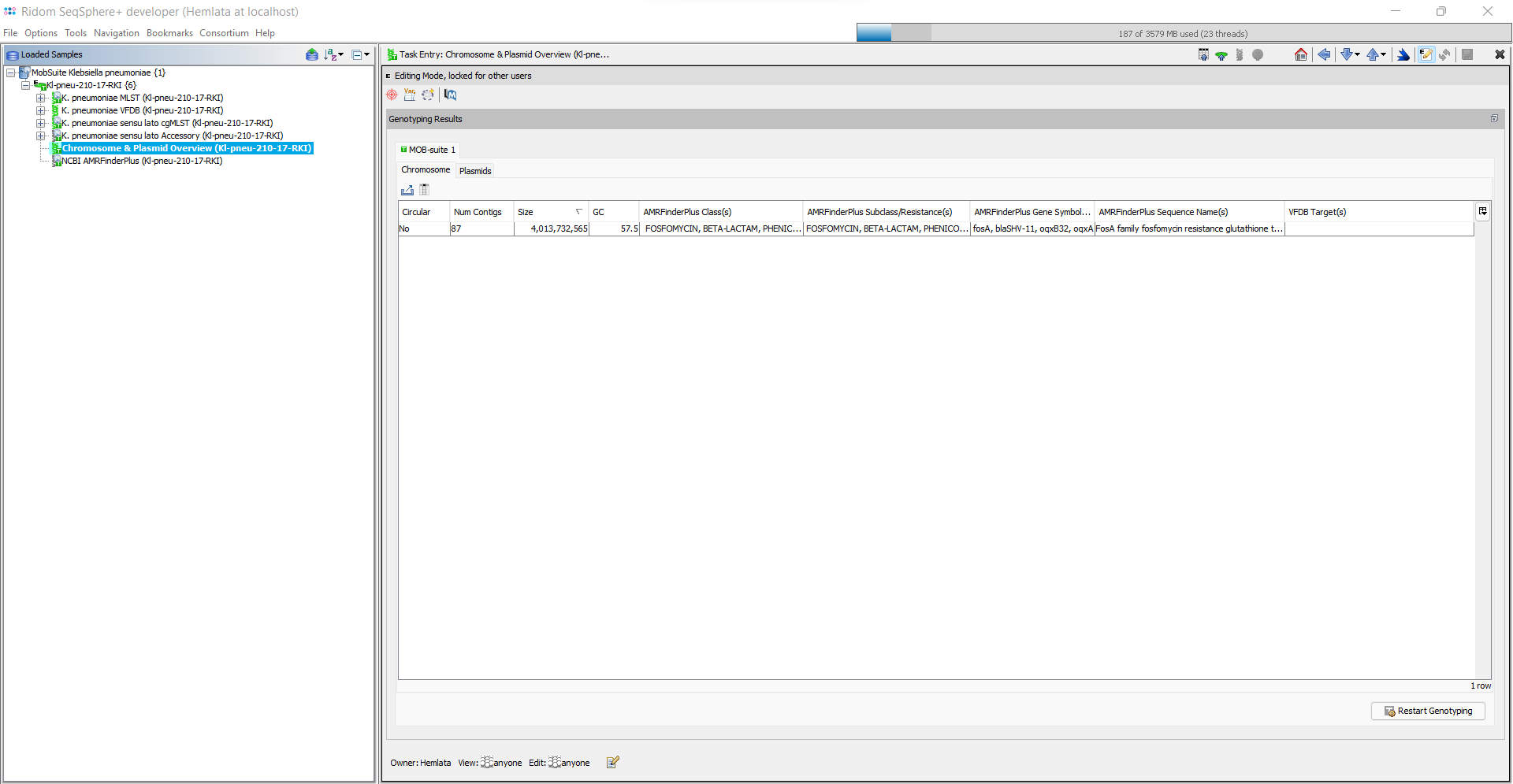
The Task Entry Overview of the processed task entry contains the two tab panels, Chromosome and Plasmids, each with an result table. By default the Plasmids table is shown. Each plasmid is listed in a separate row in the plasmid table. In the chromsome table each circular chromosome contig is listed in a separate row. All non-circular chromosome contigs are combined in one row.
The following columns exist in the two tables (but only some are shown by default):
- Molecule Designation: either plasmid or chromosome
- Plasmid Name: A plasmid name given to this plasmid. If circular it contains an order number beginning with largest plasmid. If not circular contains the size in kBases of the plasmid.
- Circular: is 'yes' if the contig name contains the text '[topology=circular]'
- Num Contigs: Number of contigs that were found for this chromosome/plasmid.
- Size: Total size of all contigs of this chromosome/plasmid.
- GC: Total GC content for this chromosome/plasmid.
- Mash Neighbor Distance: Mash distance to best matching plasmid in MOB-suite database. To show the reliability the distance value is colored green if it smaller than 0.06, else it is colored yellow.
- Mash Neighbor Accession: NCBI accession of best matching plasmid.
- Predicated Host Range: Taxon name of convergence between observed and reported host ranges.
- Host Range Associated PMID(s): PubMed ID(s) associated with records.
- Primary Cluster ID: Primary MOB-cluster id of best matching plasmid (neighbor).
- Secondary Cluster ID: Secondary MOB-cluster id of best matching plasmid (neighbor).
- AMRFinderPlus Priority Targets: AMRFinderPlus priority targets that are located on the contig(s) of this chromsome/plasmid.
- AMRFinderPlus Targets: AMR targets that are located on the contig(s) of this chromosome/plasmid.
- iMGE(s) Priority AMR: Combined identifiers for iMGE(s) that contain priority AMR target(s).
- iMGE(s) Priority AMR Accessions: Accessions of iMGE(s) that contain priority AMR target(s).
- VFDB Targets: Reliable VFDB targets that are located on the contig(s) of this chromosome/plasmid.
- Rep Type(s): Replion type(s).
- Relaxase Type(s): Relaxase type(s).
- Predicted Mobility: Mobility prediction for the plasmid (Conjugative, Mobilizable, Non-mobilizable)
- Mpf Type: Mate-Pair formation type.
- Orit Type(s): Origin of transfer type.
- Mash Neighbor Identification: Host taxonomy of the plasmid database match.
- Observed Host Range NCBI Name: Taxon name of convergence of plasmids in MOB-suite plasmid DB.
- Reported Host Range Lit Name: Taxon name of convergence of literature reported host ranges.
- Predicated Host Range Rank: Taxon rank of convergence between observed and reported host ranges.
- Observed Host Range NCBI Rank: Taxon rank of convergence of plasmids in MOB-suite plasmid DB.
- Reported Host Range Lit Rank: Taxon rank of convergence of literature reported host ranges.
- Internal sequence ID
- Contig ID: The contig name(s) that constitute a chromosome/plasmid.
- Md5: md5 hash of MOB-Suite.
- Rep Type Accession(s): Replicon sequence accession(s).
- Relaxase Type Accession(s): Relaxase sequence accession(s).
- Mpf Type Accession(s): Mate-Pair formation sequence accession(s).
- Orit Accession(s): Origin of transfer sequence accession(s).
The button ![]() can be used to export the contig sequence(s) for the selected chromosome/plasmid.
can be used to export the contig sequence(s) for the selected chromosome/plasmid.
Result Fields
The task entry stores the following result fields that can be search:
- Plasmids Found
- Plasmid Names
- Plasmid Sizes
- Primary Cluster ID
- Secondary Cluster ID
- Rep Type(s)
- Relaxase Type(s)
- Plasmid AMRFinderPlus Targets
- Plasmid AMRFinderPlus Priority Targets
- Plasmid iMGE(s) Priority AMRs
- Plasmid VFDB Targets
- Chromosome AMRFinderPlus Targets
- Chromosome AMRFinderPlus Priority Targets
- Chromosome iMGE(s) Priority AMRs
- Chromosome VFDB Targets
Except to the first field and the three Chromosome fields, the fields contain the concatenated results for each plasmid separated with an "|". If no result is available for a plasmid, a "-" is added instead.
Example:
Primary Cluster ID = AE541 | AA006 | AB561
Relaxase Type(s) = MOBP | MOBP | -
The fields can simply be searched by in the database with the "contains" operator.
If a combination of results for a specific plasmid is searched, the special criteria operator "same plasmid contains" can be chosen. This requires that two or more plasmid fields are searched together and that they are combined with AND.
![]() Hint:
By default, the result fields above are not available when creating a new Comparison Table. To change this, open the Chromosome & Plasmids Overview task template for editing and go to the result fields section. There set the flag "Use in Comparison Table by Default" for the fields that should be shown in a new Comparison Table.
Hint:
By default, the result fields above are not available when creating a new Comparison Table. To change this, open the Chromosome & Plasmids Overview task template for editing and go to the result fields section. There set the flag "Use in Comparison Table by Default" for the fields that should be shown in a new Comparison Table.
Recovering Plasmids in the Absence of Long-Read Sequencing Data
In 2021 Dutch microbiologist evaluated the performance of several plasmid reconstruction tools with short-read (Illumina) sequencing data and compared the results against the known finished plasmids of the isolates (Paganini et al. Microorganisms 9: 2021). MOB-recon showed the overall best performance. However, the authors concluded that even with this tool plasmid reconstruction from short-read data remains challenging. More specific the following problems were noted for MOB-recon:
- problems with small plasmids (plasmidSPAdes was performing here better),
- problems with plasmids carrying antibiotic resistance genes (all evaluated tools; due to an increased number of repetitive elements), and
- a tendency to split single plasmids into different predictions.
Runtimes
The following table contains the measured MOB-Suite (3.1.0) runtimes for MOB-recon and MOB-typer on a quad core (8 threads) desktop with 16GB RAM (using Windows Subsystem for Linux).
Illumina MiSeq Data
| Strain | Contig count | Genome Size (Mbases) | MOB-recon Runtime | |
|---|---|---|---|---|
| Chromosome | Plasmid | |||
| Klebsiella pneumoniae 210-17 | 87 | 8 | 5.3 | 1m |
| Ochrobactrum anthropi DSM6882 | 69 | 34 | 5.6 | 2m 7s |
| Escherichia coli 24-16 | 108 | 29 | 4.9 | 1m 15s |
| Staphylococcus aureus COL | 35 | 1 | 2.8 | 52s |
PacBio Data
| Strain | Contig count | Genome Size (Mbases) | MOB-typer Runtime (for plasmid contigs only) | |
|---|---|---|---|---|
| Chromosome | Plasmid | |||
| Klebsiella pneumoniae 210-17 | 1 | 3 | 5.4 | 33s |
| Ochrobactrum anthropi DSM6882 | 2 | 5 | 5.2 | 34s |
| Escherichia coli 24-16 | 1 | 6 | 3.6 | 44s |
| Staphylococcus aureus COL | 1 | 1 | 2.8 | 30s |