

Contents
1 Overview
The Target Parameters of a Task Template are an important feature of SeqSphere+. They are used configure the sequence processing and analysis algorithms for the target of the Task Template.
Each target of a Task Template has its own Target Parameters. However, they can also be edited all at once. But in some cases it makes sense to change the parameters for a specific target.
Some parts of the Target Parameters are only available for Sanger sequencing data Task Templates. Therefore following sections are marked with:
 - Available for Sanger sequencing data
- Available for Sanger sequencing data
 - Available for whole genome sequencing data (assembled genome data)
- Available for whole genome sequencing data (assembled genome data)
2 Base and Quality Calling
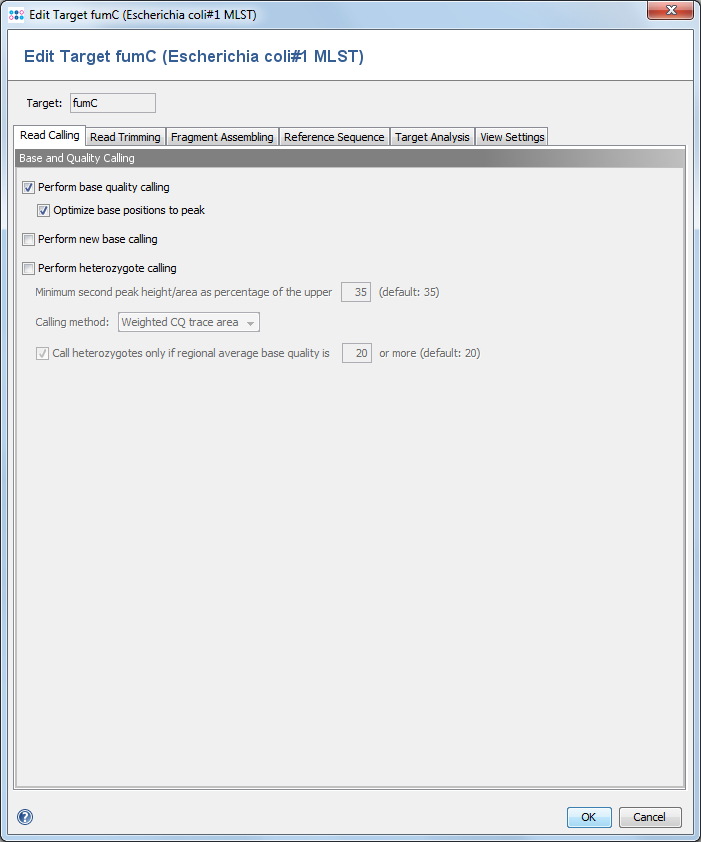
This dialog defines, if SeqSphere+ should perform quality/base/heterozygote callings on all chromatograms that are added to a target.
![]() This feature is only available for Sanger sequencing data.
This feature is only available for Sanger sequencing data.
2.1 Perform base quality calling
SeqSphere+ can calculate base qualities for each base call in the reads. The quality values are related to the error probability of the base calls. This operation will overwrite any existent base qualities that were calculated before and stored in the read files. It is recommended to enable this setting.
- Optimize base positions to peak
- Several base callers are not defining the base call position at the center of the peak, but to its upper left position. Because the Ridom base quality is calculated for the exact position, this may result in false quality values. Therefore, SeqSphere+ can correct the position of the base call to get a better quality guess for the peak. It is highly recommended to enable this option.
2.2 Perform new base calling
SeqSphere+ has a built-in algorithms to perform a base calling on chromatogram files. However, it is recommend to use the standard base-calling software (if available) that shipped with your sequencing machine, unless your results are poor.
2.3 Perform heterozygote calling
SeqSphere+ can detect heterozygote base calls in chromatograms. The heterozygote bases are called as ambiguities. The parameter for the second peak height/area can be used to configure the detection of the mixed base positions. By using the last option the heterozygote caller will be instructed to call only heterozygote bases, if a certain minimum regional (10 bases before and after the position) average base quality is surpassed.
3 Read Trimming
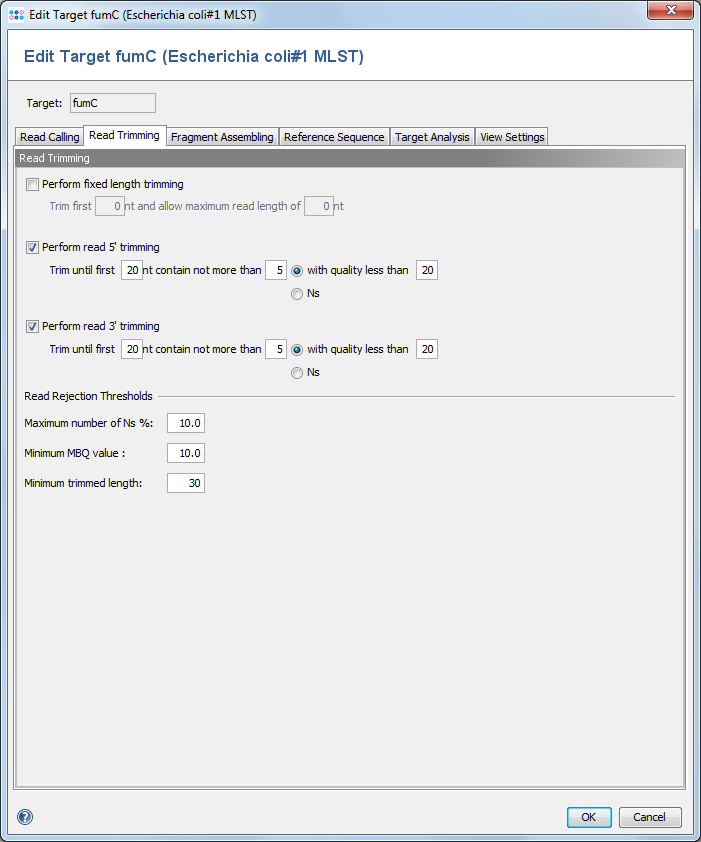
This dialog defines trimming and processing options for the reads. ![]() This feature is only available for Sanger sequencing data.
This feature is only available for Sanger sequencing data.
3.1 Perform fixed length trimming
Cuts a fixed length from the beginning of the sequence, and cuts the end of the sequence to trim it to a given length.
3.2 Perform read 5'/3' trimming
Trims the 5' and/or the 3' read region by base qualities or by occurrences of the ambiguity N. For better assembly results, it is recommended to perform at least trimmings by N's. If both trimming functions are selected, the operations will be performed consecutively.
3.3 Read Rejection Thresholds
Three thresholds can be set to define a minimum quality a read must meet to be used in an assembly. If a read does not surpass these thresholds, it will be marked as rejected in the Navigation tree. All thresholds refer to the trimmed sequence, if trimming was enabled.
4 Read Assembling
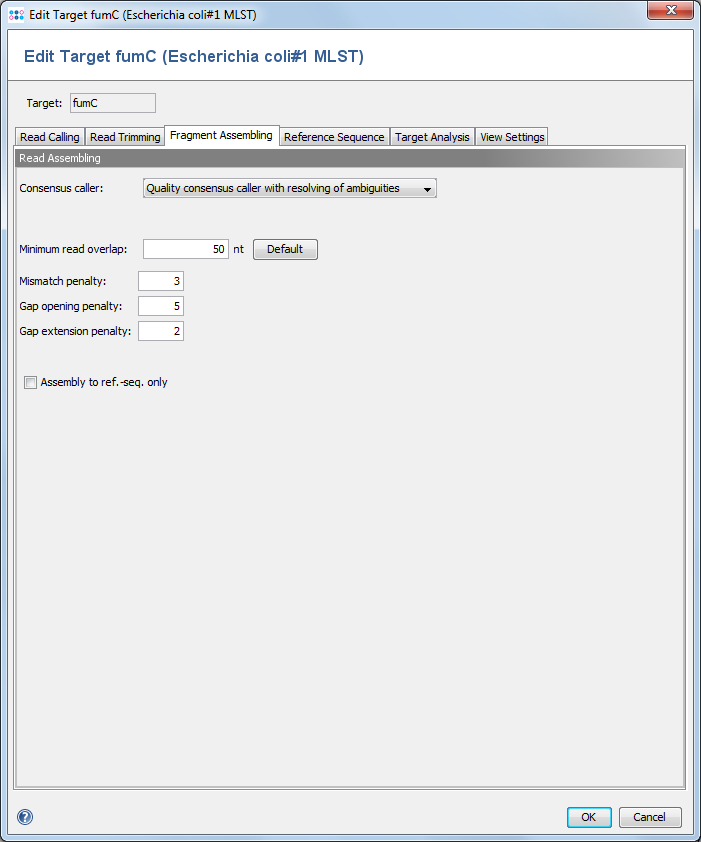
This dialogs defines the consensus calling and assembling functions. ![]() This feature is only available for Sanger sequencing data.
This feature is only available for Sanger sequencing data.
4.1 Consensus caller
The consensus caller for the contigs of an assembly can be chosen here. Currently there are five different consensus callers available.
4.1.1 Quality Consensus Caller Without Resolving of Ambiguities
This algorithm calculates the consensus base for a sequence column with Bayes formula. The quality of the read bases and the read orientation are used in this calculation. In a first step separate quality values for forward and reverse directions are calculated with Bayes formula. In a second step Bayes formula is used again to calculate a combined consensus quality value. Ambiguity bases are counted as separate base types and are not resolved. Each gap is given the quality of 20, and if the quality sum of the gaps exceeds the quality sum of all base calls, the consensus will have a gap at this position.
This consensus calling method should be used for input sequences with possible heterozygous positions (determined by a heterozygot caller) and with quality values related to an error probability by the formula <math> quality = -10 * \log_{10} (error-probability) </math>
4.1.2 Quality Consensus Caller With Resolving of Ambiguities
This algorithm is very similar to the first one (Quality Consensus Caller Without Resolving of Ambiguities). The only difference is the treatment of ambiguities: ambiguous bases are resolved here, for example a S counts as C and G.
This consensus calling method should be used for input sequences (usually with a heterozygot caller turned off) with quality values related to an error probability by the formula <math> quality = -10 * \log_{10} (error-probability) </math>
4.1.3 Majority Consensus Caller
This algorithm gives each base the same weight. The consensus base is determined by the majority base type of a column. Ambiguities cast a vote for each possible base call they represent, for example an S counts for C and G. If there is no unique majority base, an ambiguity including all bases will be called. If there are 66% or more gaps at one column position, the consensus will have a gap at this position.
This consensus calling method should be used for input sequences without quality values
4.1.4 Strict Consensus Caller
This algorithm performs a consensus calling allowing only a very limited number of mismatches. In general the algorithm performs a majority consensus call. However, all consensus columns with a coverage of only one read get a N consensus base. The table below shows the maximum allowed number of mismatches in a column for a given coverage. More mismatches will lead to a N call. All ambiguous base calls are treated as N. If there are 66% or more gaps at one column position, the consensus will have a gap at this position.
This consensus calling method should be used for forensic DNA typing
4.1.5 Inclusive Consensus Caller
This algorithm includes every base type in a column. The consensus base is the smallest ambiguity base covering all read bases. If there are 66% or more gaps at one column position, the consensus will have a gap at this position.
4.2 Minimum read overlap
This parameter can be used to configure the minimum overlap that two reads must share, before they are aligned together. Under normal circumstances the default value of 50 works fine.
4.3 Mismatch / Gap Opening / Gap Extension Penalties
The penalty values for the assembling algorithm. The defaults are 3 / 5 / 2.
4.4 Assemble to ref.-seq. only
Instead of aligning all reads to each other in an assembling process, the reference sequence only can be used for guiding the building of an assembly (this will speed up the process).
5 Reference Sequence
5.1 Reference Sequence
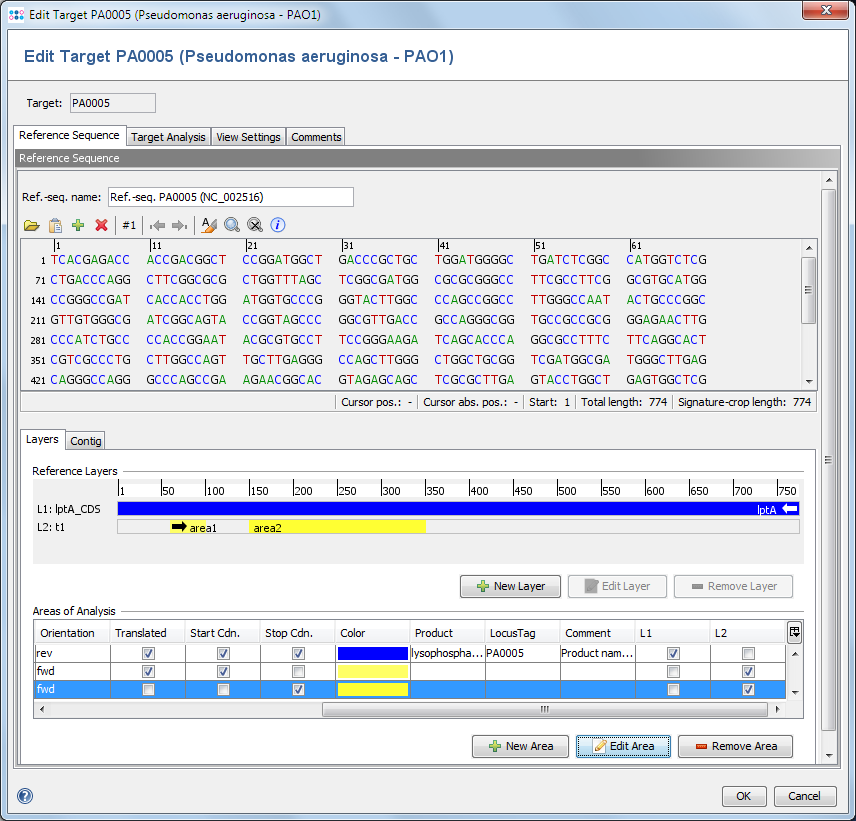
Defines a reference sequence (ref.-seq.) for the target. The main usage of a reference sequence is to orientate and crop a contig and to define variant positions. The reference sequence can be imported from a file or pasted from the clipboard.
If the Task Template was created from a seed genome, the ref.-seq. is taken from the seed core gene of this target.
5.2 Layer Settings
A layer defines a coding area that does not need to be continous. Each layer can consist of multiple areas.
5.2.1 ref.-seq. Areas
Areas are particularly labeled continuous regions in a reference sequence.
They can be imported from GenBank sequence file, or they can be created manually.
Areas are orientated (forward or reverse) and they can be marked as translatable.
Each area defines:
- Name
- Comment
- Type (e.g. gene, exon, non-coding)
- Product (from gb file)
- LocusTag (from gb file)
- Location
- Translatable
- Containing start codon / stop codon
- Orientation (forward/reverse)
- Area numbering offset (used for calculating the area Position)
- Color
5.2.2 ref.-seq. Layers
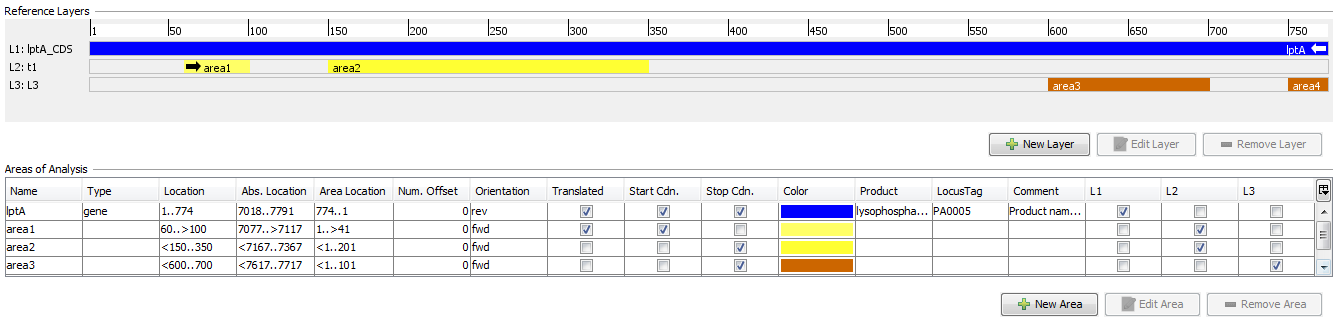
Layers are used to group one or more non-overlapping areas of a reference sequence.
By default, the first layer always covers the whole reference sequence.
Each layer defines:
- Name
- Comment
- ORF offset (+0/+1/+2)
- Orientation (forward/reverse)
- Areas belonging to this layer. All areas must have the same orientation (forward/reverse) as the layer.
5.2.3 Different Position Specifications
- Contig ref.-seq. Alignment
- A position of a base in the consensus is determined by the aligned position the reference sequence. If no reference sequence alignment can be done, the consensus starts with 1.
- Position
- This is the default position specification. The first base in the ref.-seq. is by default position 1. It can be redefined in the Task Template parameters.
- Absolute Position
- This position is useful when the ref.-seq. was extracted from a genome file. It gives the position of a base in the genome. If the ref.-seq. was not extracted, the absolute position is the same as the standard position.
- Area Position
- Gives the position of a base in an Area. It is calculated from the area number offset that is defined in the area and counted in the orientation of the area.
5.3 Contig Settings
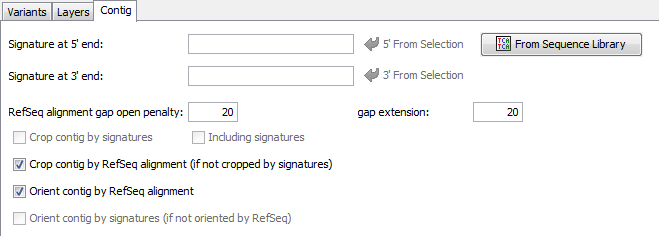
5.3.1 Contig Signatures
Defines the 5' and the 3' signature for a contig target. For convenience the signatures can be copied directly from a selection made in the reference sequence (buttons 5' and 3').
In addition the signatures can be imported from a multiple alignment file by using the From Sequence Library button. Again an inclusive consensus will be called and the beginning and the end of the consensus can be used as signatures. The Including signatures check-box can be used to define, if the signatures itself should belong to the contig, or not.
The signatures may contain ambiguities. In this case, the signatures will match to every base that is expressed by this ambiguity, and to the ambiguity itself. If the signature should not match the ambiguity but only to the different bases, the base characters must be grouped with [ ]. Example: W matches to A,T or W; but [AT] matches only to A or T.
5.3.2 ref.-seq. Alignment Settings
The gap opening penalty, and the gap extension penalty for the alignment between the consensus and the ref.-seq. can be set.
5.3.3 Contig Cropping and Orientation
The signatures and the reference sequence can be used to orientate and to trim the contig automatically.
6 Target QC Procedure
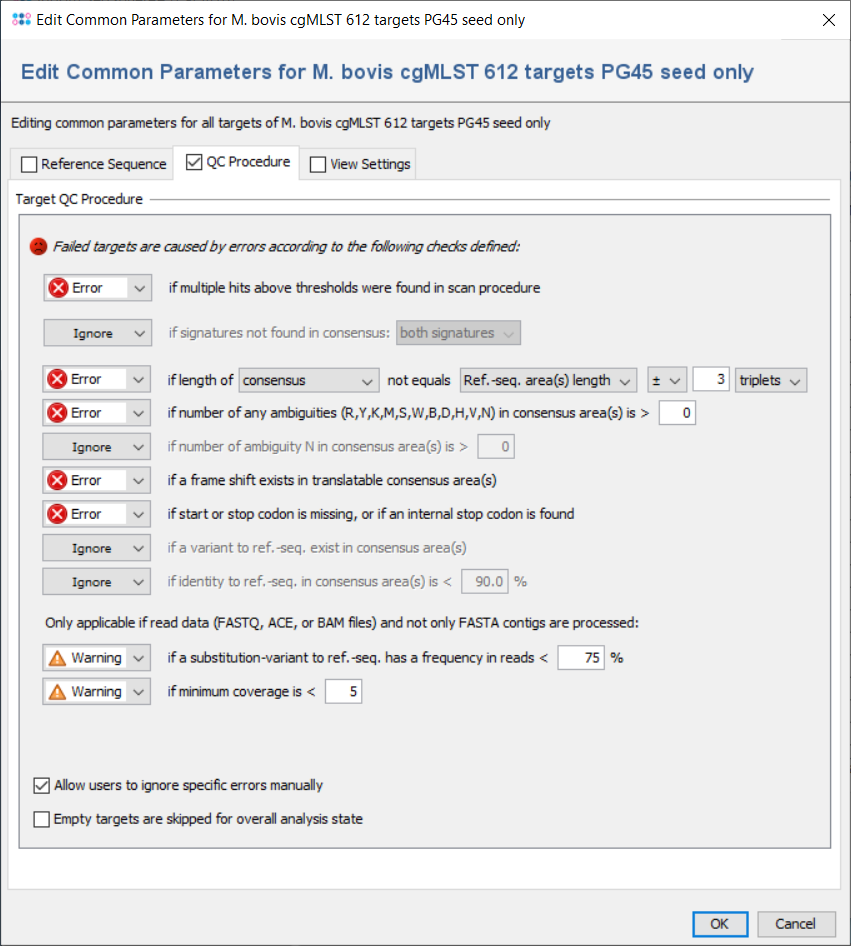
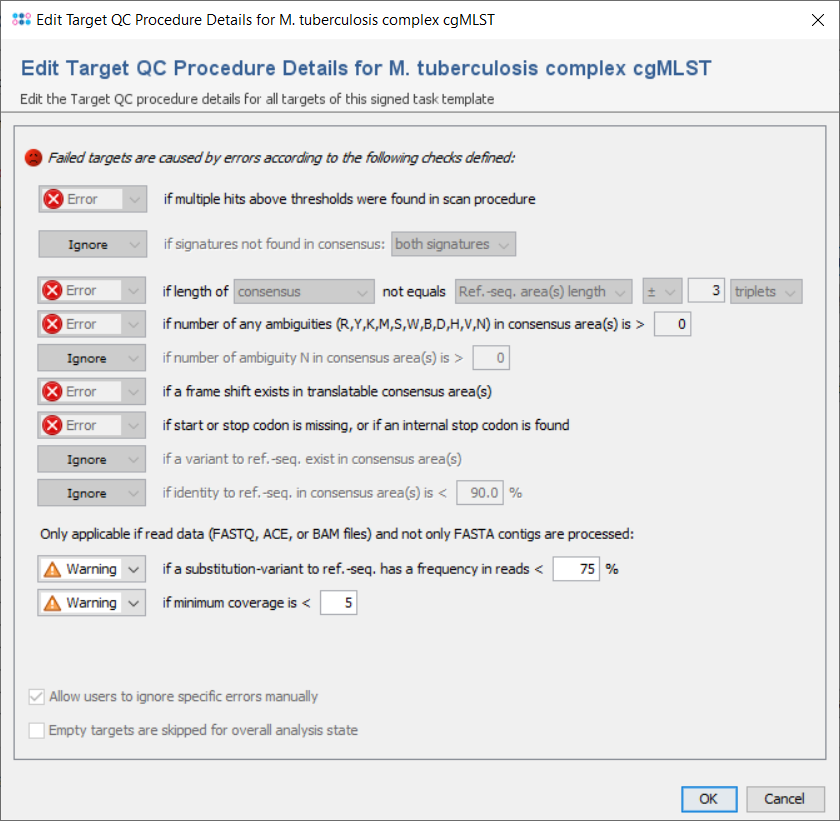
All targets are automatically checked for the quality issues that were defined in the Analysis Parameters of the Task Template. Each check can set to
- Ignore:
- Check is not done at all.
- Warning:
- If target fails the check it is rated as
 good with warnings.
good with warnings.
- If target fails the check it is rated as
- Error:
- If target fails the check it is rated as
 failed.
failed.
- If target fails the check it is rated as
But default, the parameters are the same for all targets, but they can also be defined targets-specific. If a target has multiple sequences and if this is allowed by the first check), all further checks are performed for the first one only,
The checks always work on the areas of the layers that are defined for the target in the task template. Usually (e.g., in cgMLST), a target has only one layer with a single area that covers the whole target sequence, and defines its orientation (forward/reverse). Therefore, the terms 'layer' and 'consensus area(s)' are in most cases equivalent to the target sequence.
The following checks can be set to Ignore/Warning/Error:
- if multiple hits above thresholds were found in scan procedure
- Defines if multiple hits above the identity and aligned thresholds are allowed and imported in the target scan procedure, or not.
- if signatures not found in consensus
- Defines that the 3` signature, the 5`signature, or both must be found in the consensus.
- if length of consensus not equals ref.-seq. length with a specified tolerance
- Defines that the trimmed consensus, or all bases of the consensus that are covered by a ref.-seq. area, must have a specific length, or the same length as the reference sequence in all areas of this layer. A tolerance range for the length comparison can be set in bases or codons.
- if minimum mean base quality is above a specified value
- Defines the allowed minimum mean base quality, calculated for all bases in the consensus areas of this layer.
 This feature is only available for Sanger sequencing data.
This feature is only available for Sanger sequencing data.
- if number of low quality bases is above a specified value
- Defines the allowed maximum number of low quality bases in the consensus areas of this layer. The threshold for a low quality can be specified. This feature is only available for Sanger sequencing data.
- if number of any ambiguities (R,Y,K,M,S,W,B,D,H,V,N) is above a specified value
- Defines the allowed maximum number of any ambiguities in the consensus areas of this layer
- if number of any ambiguity N is above a specified value
- Defines the allowed maximum number of the ambiguity N in the consensus areas of this layer
- if a frame shift exists in translatable consensus area(s)
- Defines that the number of insertion(s) in comparison to ref. seq. minus the number of deletion(s) must be dividable by 3.
- if start or stop codon is missing, or if an internal stop codon is found
- Defines that the translation of the consensus areas according to the layer must contain a start codon and a stop codon only at the expected positions, if they are defined in the areas of an translatable layer. Therefore, if layer and target sequence are identical as it is usually the case this check fails if no start codon is found at the begin, no stop codon is found at the end, or a stop codon is found at a wrong position in a target.
- if a variant to ref.-seq. exist in consensus area(s
- Demand that no differences exist between the consensus and the ref.-seq. in the areas of this layer.
- if identity to ref.-seq. is below a specified value
- Identity is calculated as the rational difference between consensus and reference sequence in an alignment. Differences are gaps and mismatches. Ambiguities never match to bases, but to the same ambiguity
- if a substitution-variant to ref.-seq. has a frequency in reads is below a specified value
- Defines that substitution-variants to the ref.-seq. must be confirmed by a specific percentage of reads that cover this position (Default: Warning if below 75%).
- if minimum coverage is below a specified value (Whole Genome Sequencing)
- Defines the coverage that is demanded for every base (is not average coverage) in the consensus areas of this layer in any reading direction (Default: warning if below 5). This feature is only available for whole genome sequencing data.
- if minimum coverage is below a specified value (Sanger Sequencing)
- Defines the coverage that is demanded for the bases in the consensus areas of this layer, and optionally if both reading directions (forward and reverse) should be covered in the reads. The coverage is defined by percentage for to the base count in the consensus areas. To handle badly covered ends of the contig, it can be defined that the uncovered positions (if demanded coverage <100%) may only appear on the ends of the contig. This feature is only available for Sanger sequencing data.
6.1 Processing Options
- Allow users to ignore specific analysis problems
- If this option is checked, an Ignore button is added to the analysis problem table in sequence view.
- Empty targets are skipped for overall analysis state
- By default, targets without sequence will set the analysis state of the whole task entry to 'bad'. If this option is set, any targets without sequence will be ignored during analysis and will have no effect on the analysis state of the task entry.
7 View Settings Defaults
These settings can be used to define which views should be opened initially for the contigs of the according Task Template.
7.1 Contig Alignment
- Show Quality Colors
- Initially show the base qualities as background colors in the contig alignment.
7.2 Additional Views
- Show ref.-seq. Alignment, if ref.-seq. is defined
- Initially show the alignment between the ref.-seq. and the contig, if a ref.-seq. is defined
- Show Read Sequences
- Initially show the chromatograms of the contig reads. This feature is only available for Sanger sequencing data.
7.3 Information Tables
- Show Variant Table
- Initially show the table with all variant positions between the ref.-seq. and the contig
- Show Analysis Table
- Initially show the table with the target analysis results and warnings
7.4 Task Entry Overview
- Maximize Query Result Panel
- Initially fills the whole Task Entry overview with the query result panel.