

Contents
Overview
SPAdes (version 4.2.0, citation) is a de novo assembler for whole genome read data that can be used in the Ridom Typer assembling pipeline if the software is running on Linux. As SPAdes is not available for Microsoft Windows it is only delivered with the Linux version of the Ridom Typer Client. SPAdes works well for assembling reads from Illumina systems or from Ion Torrent machines.
Quality Trimming
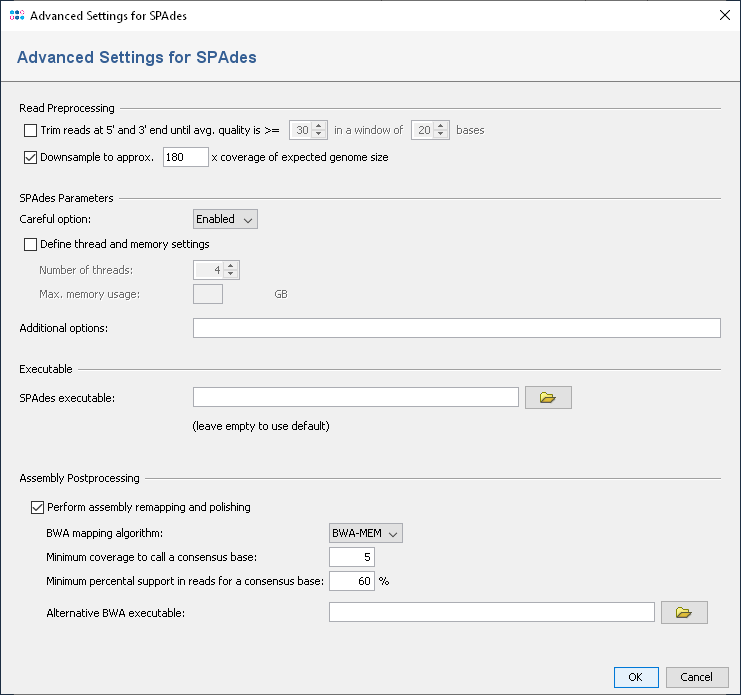
SPAdes performs an error correction of the read data. Therefore it is not recommended to use the Ridom Typer read trimming as pre-processing (disabled by default).
Downsampling
To reduce the size of the output files and time and memory usage, the input files can be downsampled. Downsampling randomly removes reads so that the given approx. size is obtained. If quality trimming is selected, downsampling is done on the trimmed reads.
Depending on sequencing technology and read length different downsampling settings are useful. For Illumina MiSeq data downsampling to 180x is suggested (enabled by default). However, downsampling can be disabled if wished.
Remapping
By default a remapping is performed in a pipeline that uses SPAdes with the default settings. After the SPAdes process has finished, BWA is used to align the reads against the resulting assembly contigs. BWA mem is used as algorithm and for the resulting alignment (BAM file) a new consensus is called. As SPAdes is known to create inacurrate alignments for lower coverage, the consensus calling procedure is configured to use a threshold of 5 as minimum coverage to call a non-ambiguous base. The required read support for a consensus base is 60%.
SPAdes Options
An automatic k-mer selection of SPAdes based on sequence length (e.g., 21,33,55,77 for 150bp and 21,33,55,77,99,127 for 250bp) is used for the assemblies by default. However, this can be changed by input of command line parameters in additional options.
The "--careful" option performs a mismatch correction. This option is enabled by default in the pipeline.
If the Sequencing Platform of the pipeline is set to Ion Torrent, the "--iontorrent" option is automatically added.
The number of threads and maximum memory usage can be specified. If nothing is specified, the SPAdes default is used (16 threads).
Furthermore, additional command line parameters for SPAdes can be defined (e.g., the k-mer selection).
If SPAdes was used as assembler in a pipeline, then the logging output of SPAdes can later be viewed in the Procedure tab of the finished Sample.
SPAdes Version
If no path is given, the SPAdes executable (spades.py) is searched in the subdirectory ext/ of the Ridom Typer Client installation directory. To update the SPAdes version, the full spades installation directory could copied in the ext/ directory, after removing the old one.
However, Ridom Typer is only tested with the SPAdes version that is delivered with the software.
Installation Issues
On Windows, SPAdes requires WSL version 2.
On Linux, SPAdes requires that the python interpreter is available under the name 'python' (and not only as python3). Therefore, e.g., on Ubuntu 22.04 it is necessary to install the debian package python-is-python3.
FOR RESEARCH USE ONLY. NOT FOR USE IN CLINICAL DIAGNOSTIC PROCEDURES.