

Contents
Prerequisites
![]() Important:
In the MinKNOW software it is necessary that the option Trim barcodes is set to ON for the MinKNOW experiment.
Important:
In the MinKNOW software it is necessary that the option Trim barcodes is set to ON for the MinKNOW experiment.
MinKNOW Run Directory
MinKNOW stores each run in a directory named by the run ID. The fastq_pass directory in it contains for each barcode a subdirectory with multiple FASTQ files. When processed with the Ridom Typer pipeline, they will be merged into one FASTQ file per barcode and stored in the specified output directory. The run directory also contains the file 'report_<run-id>.json' in the run directory that stores detailed information about the run. This report file is required for this Ridom Typer pipeline and is automatically imported as run details.
For assembling the MinKNOW data, Ridom Typer requires
- the fastq_pass with the barcode directories
- the report json file
If the MinKNOW run directory is accessible from the Ridom Typer client, it can directly be selected as input directory. To process multiple runs at once, the directory above the run directories can be selected.
Alternatively, if the MinKNOW directories are not directly accessible, it is also possible to copy the fastq_pass directory and the report json file into a new directory, and use this directory as input directory of the pipeline.
Pipeline settings
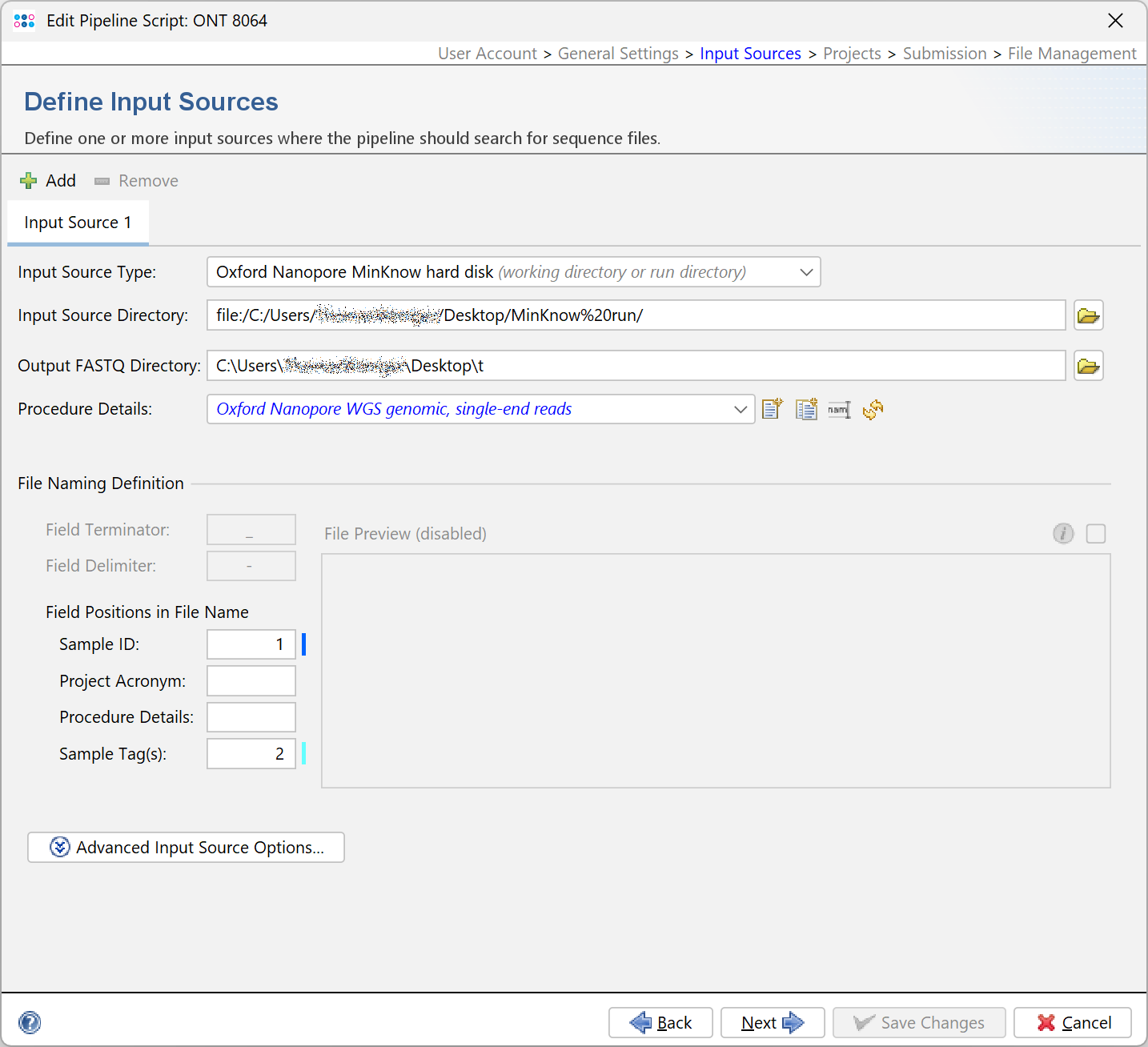
Selecting Oxford Nanopore MinKNOW hard disk as Input Source Type allows to import all samples from a MinKNOW run directory using a pipeline script.
When Oxford Nanopore MinKNOW hard disk is selected as Input Source Type an additional input field is available: Output FASTQ directory.
The FASTQ-files for a MinKNOW sample are merged together and stored in a subdirectory of Output FASTQ directory that is named by the run id, e.g. 20241209_1142_MN30839_FBA87915_d4fdb8bb. Therefore this directory must be writeable for the current user.
Note that the field Field Terminator cannot be edited when Oxford Nanopore MinKnow hard disk is selected and that the file preview is disabled.
Running the pipeline
A subdirectory named by the run ID is created in the output FASTQ directory. Merged FASTQ files, the report file and a sequence specification file (sequence_specification.spec) are written to this subdirectory.
FASTQ files for a run that already has a subdirectory below the output FASTQ directory will be ignored by the pipeline.
Sample Sheet
A sample sheet is used to assign a sample name to the barcodes. The sample sheet is stored in a file sample_sheet_ridom_typer.csv or sample_sheet_ridom_typer.xlsx in the ONT run directory.
This file can be created manually or by using the sample sheet input window.
The sample sheet file must contain at least two columns (with headers). The first column is used for barcodes (e.g. barcode01 ... barcode99), the second column contains the sample name. All other columns are ignored. The column headers can contain any name. The sample name may only contain the characters a-z, A-Z, 0-9, <Space>, and -.

Sample Sheet Input Window
If a ONT run directory does not contain a file sample_sheet_ridom_typer.csv or sample_sheet_ridom_typer.xlsx and no run subdirectory below the output directory exists a sample sheet input window is opened when the pipeline is started. The window can contain several tabs if the sample data has to be entered for several runs.
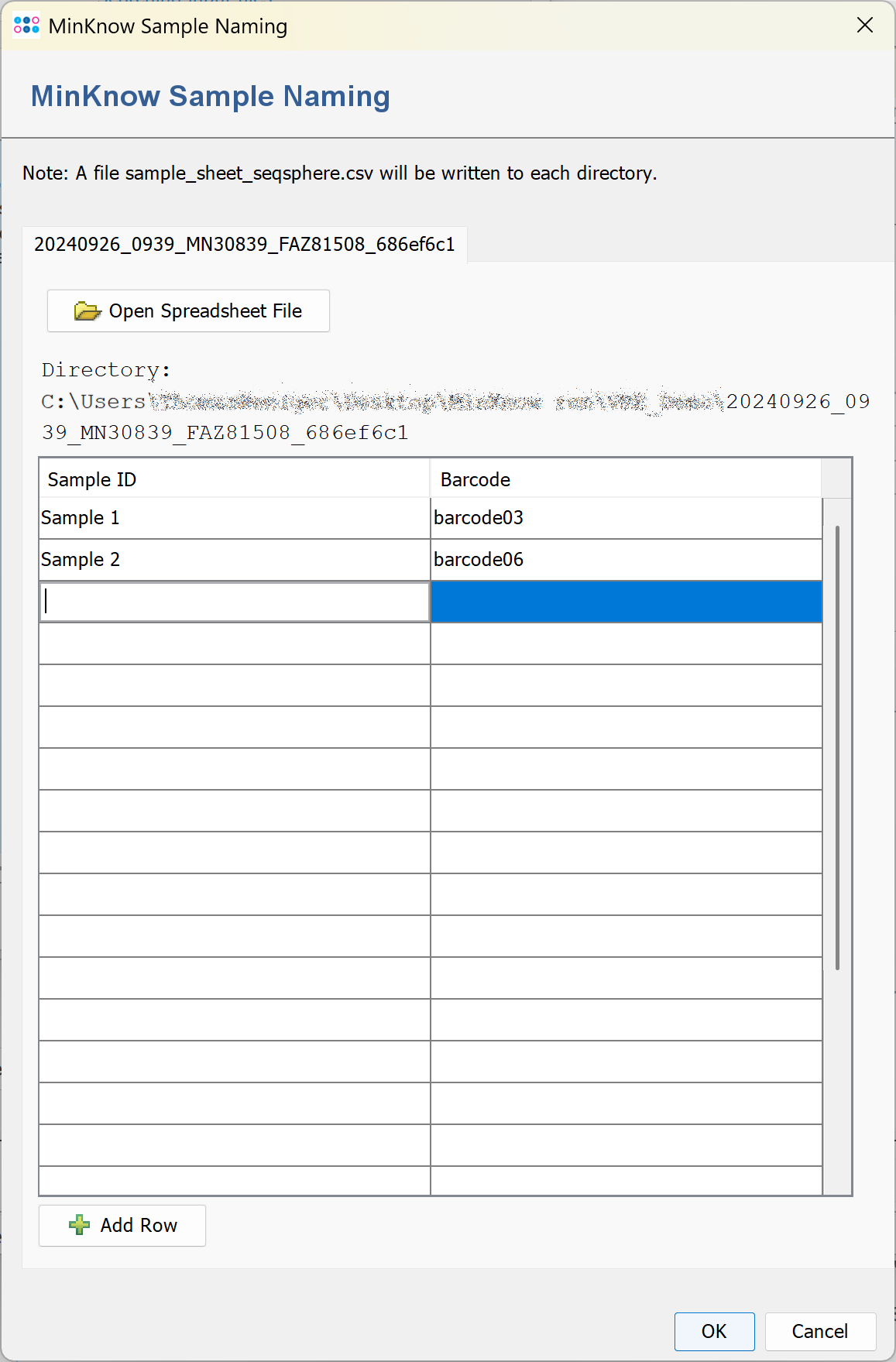
Enter the Sample names and choose the corresponding barcodes. The sample name may only contain the characters a-z, A-Z, 0-9, <Space>, and -.
After clicking OK in the sample sheet window the file sample_sheet_ridom_typer.csv is written into the ONT run directory. The run directory must be writable by the Windows user for this to work.
If the sample sheet input window is open and the file sample_sheet_ridom_typer.csv in the run directory is written by another computer (e.g. on a network drive) the sample sheet for this run is removed from the window. If there are no more sample sheets to enter for the other directories, the window will close.
![]() Hint: If multiple barcodes belong to a single sample, the same sample name can be entered multiple times in the sample_sheet_ridom_typer-file or in the sample sheet input window. The FASTQ files from all specified barcode directories are then merged into one file.
Hint: If multiple barcodes belong to a single sample, the same sample name can be entered multiple times in the sample_sheet_ridom_typer-file or in the sample sheet input window. The FASTQ files from all specified barcode directories are then merged into one file.
Pipeline in Continuous Mode
Using a pipeline in continues mode (see page Pipeline Script for details), further processing of the data generated by MinKnow can be started together with the sequencing.
- Start Sequening in MinKnow. Set Trim barcodes to on.
- Start a Ridom Typer pipeline (with Oxford Nanopore MinKNOW hard disk as input source type) whose input directory corresponds to the MinKnow data directory.
- Shortly after MinKnow has written the first basecalling results, the sample sheet input window will open. Enter the assignment of the barcodes to the sample names and press OK.
- Shortly after MinKnow has finished sequening and basecalling, the pipeline will start importing the samples.
Procedure Details
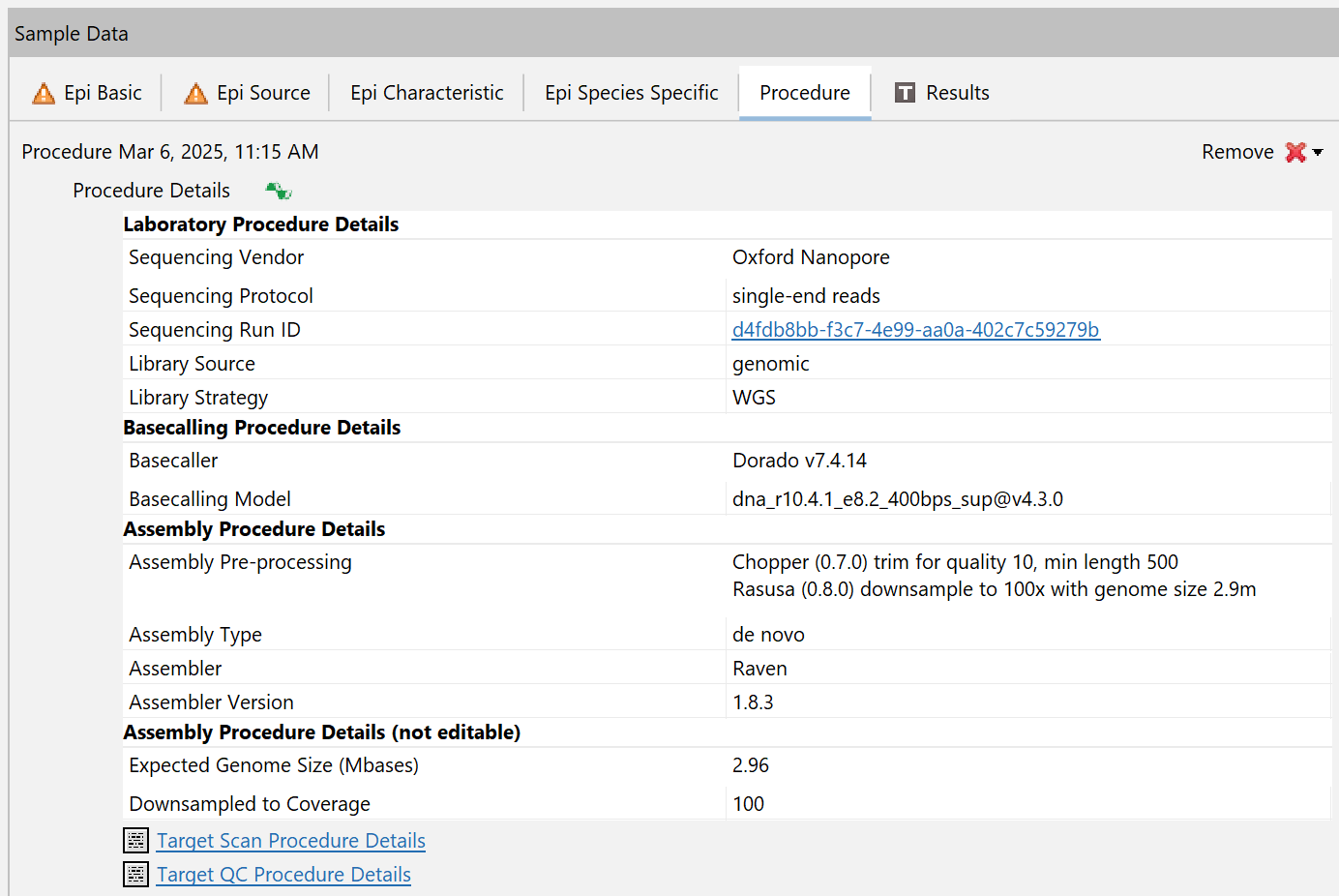
Information about the MinKNOW run are stored an a Sample's procedure details, e.g. the used basecaller and assembler. Information about the sequencing run can be accessed by clicking the cell with the Sequencing Run ID.
FOR RESEARCH USE ONLY. NOT FOR USE IN CLINICAL DIAGNOSTIC PROCEDURES.