

General
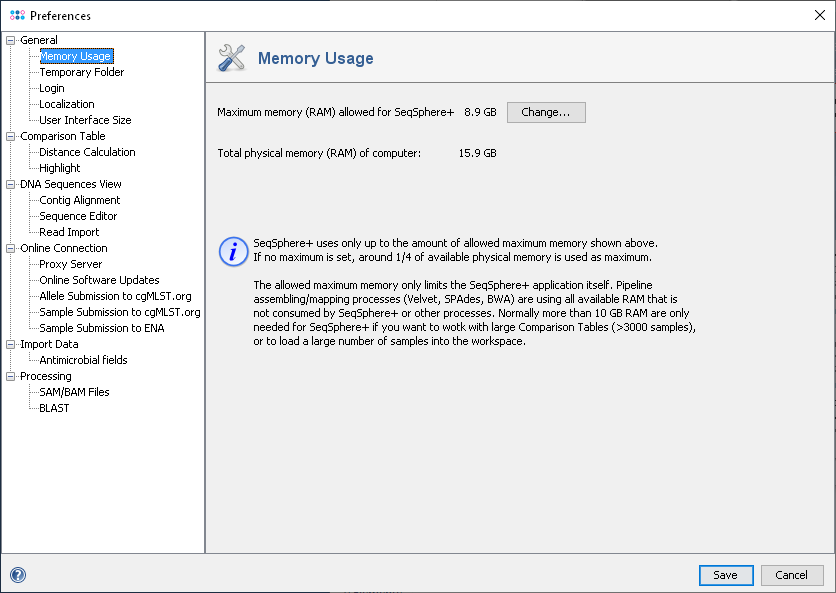
- Memory Usage
- Configure the maximum of memory (RAM) that can be used by Ridom Typer. By default 25% of the physical memory are used a minimum of 8 GB is recommended.
Assemblers that are called in the pipeline are running in an external process and are therefore they not limited by this maximum. However, there must be enough physical memory left available for those external processes. Increase the maximum of memory (RAM) that can be used by Ridom Typer above the default value only if you want to process or load a large number of Samples or if you want to create very large Comparison Tables.
- Temporary Folder
- Allows to change the temporary folder that is used by Ridom Typer for all temporarily created files (e.g., for large temporary assembling data).
- Login
- Allows to disable showing the last used login name as default in the login dialog (e.g., if the Ridom Typer client is used by multiple users).
- Localization
- Allows to set some country specific defaults.
- User Interface Size
- Allows to increase the size of fonts and button icons.
Comparison Table
- Distance Calculation
- Allows to hide the warning dialog for columns with empty values
- Highlight
- Allows to disable the highlighting of empty values
DNA Sequence Views
- Contig Alignment
- You can specify if the visible layout of the aligned read rows when scrolling through the contig alignment.
- Sequence Editor
- You can specify the color schemes visible layouts for the sequence and chromatogram editors
- Read Import
- You can specify if the name of a read should be take from the file name, or from an internal comment
Online Connection
- Proxy Server
- The Proxy settings must be specified here if your need to go through a HTTP Proxy to access the Internet.
- Online Updates
- Configure if Ridom Typer should search for updates on every startup.
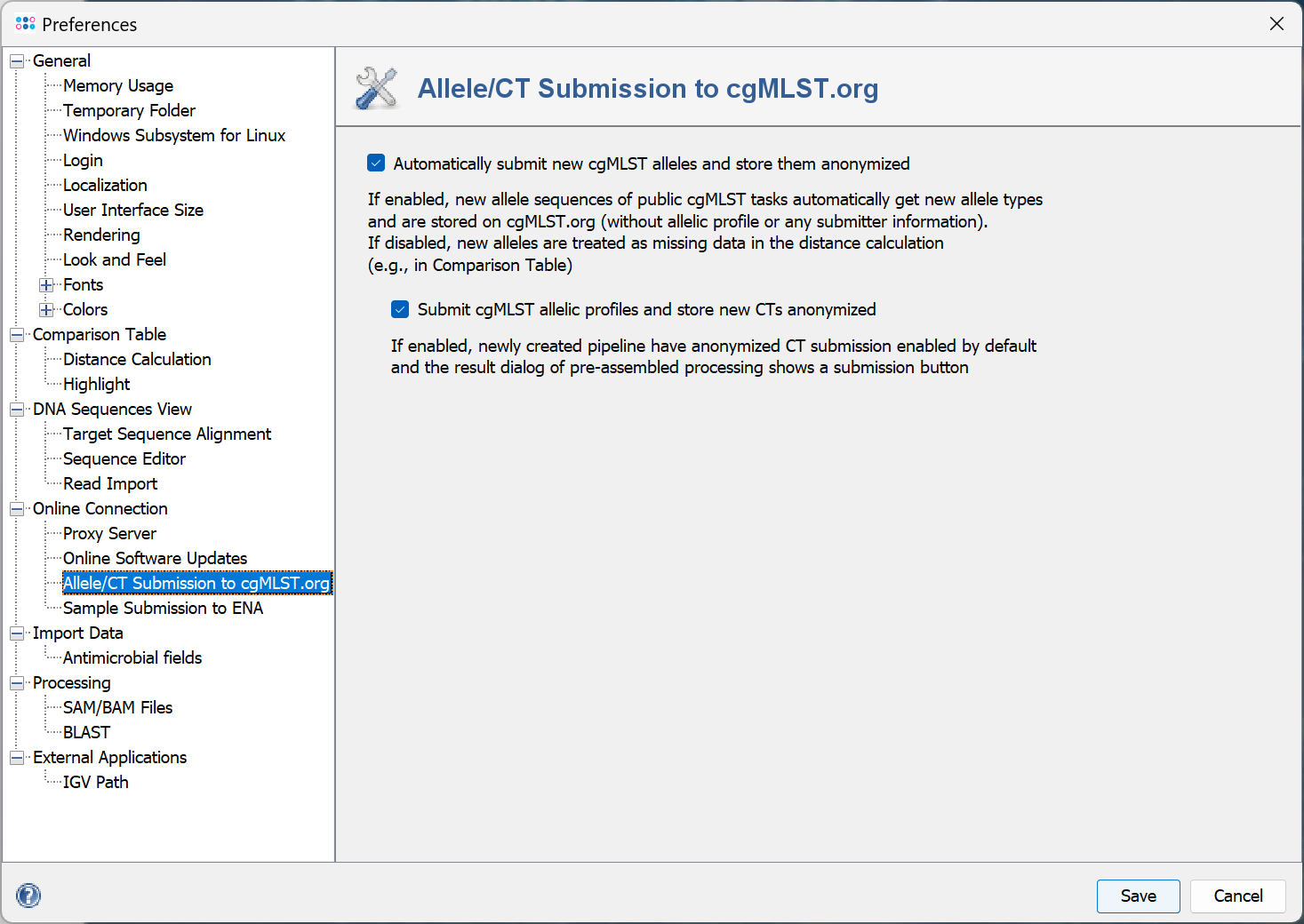
- Allele/CT Submission to cgMLST.org
- The allele submission box defines if new allele sequences of public cgMLST tasks should automatically get new allele types. If enabled they are stored on cgMLST.org (without allelic profile or any submitter information). If disabled, new alleles are treated as missing data in the distance calculation (e.g., in Comparison Table).
- The allelic profile and CT submission box defines if newly created pipelines have the anonymized CT submission enabled by default and that in the result dialog of pre-assembled processing a submission button is shown.
- See Submission to cgMLST.org for more details.
- Sample Submission to ENA
- Allows to specify the default Submission Anonymization Filter to define what level of place/time and sample ID should submitted to ENA database. Submission to ENA is never performed automatically, but must be manually invoked. The filter that is defined here will be used as default when opening the submission dialog.
Input Files
- SAM/BAM
- BAM are produced by SKESA/SPAdes pipelines with de novo remapping enabled, and by BWA pipelines.
- Ridom Typer can also import SAM/BAM files from other sources.
- The following settings can be specified here for reading SAM/BAM files:
- Discard reads with a mapping quality lower than a defined threshold (default 10)
- Ignore illegal reads (e.g., bad cigar string). If this option is disabled, the processing of the BAM file aborts with an error if illegal reads are found reads that are marked to have more than one best hit (X0 tag > 1) or as secondary alignment (bitwise FLAG)
- Discard reads that are marked as PCR or optical duplicate (bitwise FLAG)
FOR RESEARCH USE ONLY. NOT FOR USE IN CLINICAL DIAGNOSTIC PROCEDURES.