
| wiki | search |
Velvet is a de novo assembler for short read data. Velvet works well for assembling reads from Illumina systems with read length from 70bp to 250bp length. However, Velvet does not work well with reads that contain many InDel-errors and is therefore not suited for assembling data from Ion Torrent or 454 machines.
The assembler's algorithm is based on a so-called 'k-mer' data structure. The assembler results strongly depend on the chosen value for k. An automatic mode is included in SeqSphere+ that runs Velvet for multiple k-values and uses the results from the best run. Multiple instances of Velvet can be run in parallel with different values of k to speed up the assembling. Velvet can require large amounts of memory, therefore at least 32GB of memory are recommended.
The Velvet assembler can be accessed using the menu Tools | Short Read Assembler (Velvet).
The assembler reads from FASTQ-files that contain either single or paired reads. The input files can be quality trimmed and downsampled before assembling. The assembled reads are written into an ACE-file, they can be imported using Create Samples from Assembled Genomes.
Contents |
Quality Trimming
The reads are processed before they are assembled. They can be automatically trimmed based on read quality and downsampled.
Default settings for trimming are to trim on both ends of the reads until the average base quality is > 30 in a window of 20 bases. This settings usually work well for Illumina HiSeq/MiSeq data. For other sequencing technologies different values might be used. Note that quality trimming will not remove a read with quality below threshold in all bases, but will always leave (2*window size) bases in the middle of the read.
It is recommended to enable quality trimming, it usually results in better assemblies.
Downsampling
To reduce the size of the output files and time and memory usage, the input files can be downsampled. Downsampling randomly removes reads so that the given approx. size is obtained. If quality trimming is selected, downsampling is done on the trimmed reads.
Depending on sequencing technology and read length different downsampling settings are useful. For Illumina HiSeq data downsampling to approx. 125x the expected genome size worked well, for Illumina MiSeq data downsampling to 180x is suggested. On a fast system with much memory and when file size is not a problem downsampling can be disabled.
Choose of k-values
Velvet uses a 'k-mer' data structure for assembling. Assembling results differ based on the value for k. A good value for k can be vaguely guessed from the coverage and the read length. To get good assembling results, it is suggested to run Velvet using different values for k and return the assembly with the best results (measured by avg. length of contigs with more than 1000bp).
An automatic mode exists that uses an heuristic based on read length to determine k-values that should be checked. Velvet is then run for these k-values. The automatic mode usually finds good results, and using it is recommended for most users. Alternatively, an upper and a lower limit for k-values and a step size can be specified. Velvet is then started for all k-values in the given range and the best assembly is returned.
Automatic mode heuristic
- Start on the lowest value of
- average read length/2
- 107
- End at the lowest value of
- average read length * 0.8
- 171
- Search first using k-mer step size of 8, then around the best hit with step size 2.
Settings
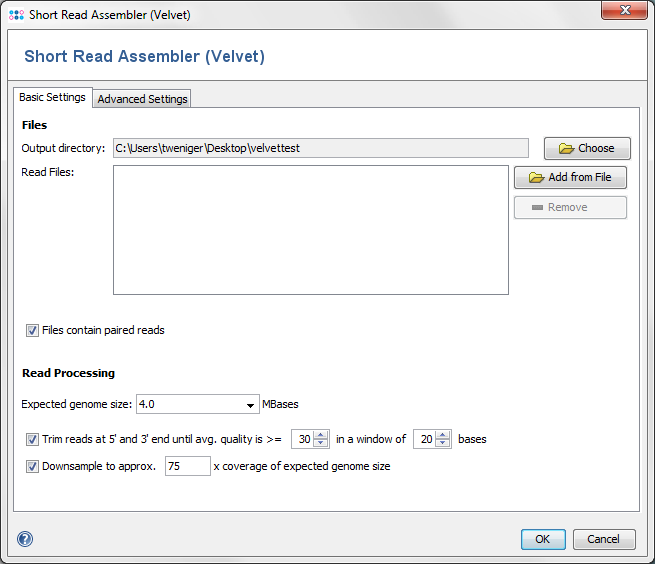
The dialog window allows to change settings for
- Output directory: the directory where the resulting ACE-files will be written to.
- Read Files: the read files, in FASTQ or FASTQ.GZ format. Multiple files can be selected, and they can be automatically grouped by their filename (e.g. forward and reverse reads can be grouped together).
- Paired Reads: Check if the files contain paired reads. The assembler uses paired reads if exactly two files are in each file group. Both forward and reverse files must contain the same amount of reads, and read number X in the forward file must correspond to read number X in the reverse file.
- Expected genome size: The expected size of the assembled genome (for downsampling).
- Quality trimming: reads can be trimmed from both ends until their quality is above the given average quality within a given window.
- Downsampling: select which coverage of expected genome size should be reached by downsampling.
- k Range: The minimal value for k, the maximum value and the step can be specified. Note that the k-values must be odd, and the step must be even. When automatic mode is used, k-values to check are calculated automatically.
- Optimize coverage cutoff: Do coverage cutoff optimization for each Velvet run. This usually improves the resulting output.
- Simultaneous Velvet Runs: The number of simultaneous runs can be specified. Note that running multiple instances of Velvet in parallel will result in higher memory consumption. See paragraph Speed and Memory.
Speed and Memory
Memory usage of Velvet depends on
- number of reads
- read length
- read qualities
- read distribution (depends amongst others on used chemistry)
- genome size
- k-value
If possible, quality trimming and downsampling is recommended to reduce memory consumption. When running multiple instances of Velvet in parallel, memory consumption increases.
| Genome size | Coverage | Read length | Runtime | Used Memory for single Velvet run |
|---|---|---|---|---|
| 2.8 MBases | 131x | 150bp | 15min | ~1GB |
| 2.8 MBases | 150x | 250bp | 21min | ~1.6GB |
| 5.5 MBases | 150x | 150bp | 22min | ~2GB |
| 5.5 MBases | 150x | 250bp | 43min | ~5GB |
| 6.2 MBases | 67x | 150bp | 18min | ~2GB |
| 6.2 MBases | 150x | 250bp | 66min | ~8GB |
Note that actual time and memory requirements may be different depending on genome size, read length, coverage, read distribution and quality, and k-mer settings.
The table allows to estimate how many instances of Velvet can be run on parallel. For example, on a system with 32GB, up to 4 instances of Velvet can be run in parallel for a genome size of up to 6.2 MBases using downsampling to 150x and default clipping. For smaller genomes, a higher number of instances can be run in parallel if enough processor cores are available.
Open Source note
The Velvet assembler function is a wrapper to external Velvet executables. The Velvet software is open source software that is licensed under the GNU General Public License version 2.0 (GPLv2). The program homepage is http://www.ebi.ac.uk/~zerbino/velvet/. The program was ported to windows by Applied Maths: http://www.applied-maths.com/download/open-source. The source code is installed next to the executable.
Please note that Ridom can only give limited support for the Velvet assembler.
For more information see: Zerbino DR and Birney E (2008) Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18(5):821-9. [PMID: 18349386]