

Overview
SKESA (citation) is a de novo assembler for Illumina whole genome read data. It can be used in the assembling pipeline if the SeqSphere+ client is running on Linux or if the Windows Subsystem for Linux is installed on Windows.
SKESA is De Bruijn graph-based and designed for assembling reads of microbial genomes sequenced using Illumina. It is not suited for assembling data from Ion Torrent or 454 machines.
See the page SKESA Evaluation for a detailed comparison of the performance of SKESA, SPAdes, and Velvet.
Quality Trimming
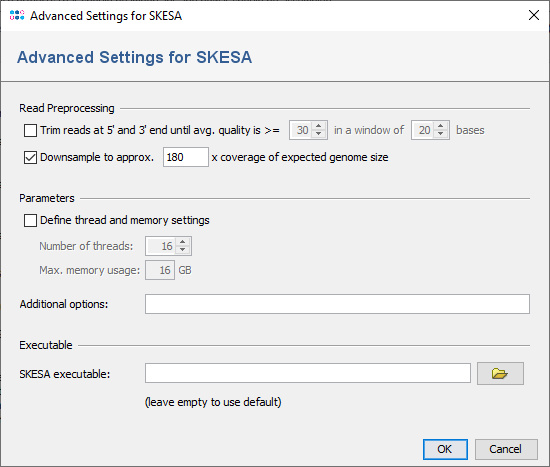
SKESA performs a trimming of the read data. Therefore, it is not recommended to use the SeqSphere+ read trimming as pre-processing (disabled by default).
Downsampling
To reduce the size of the output files and the time and memory consumption, the input files can be downsampled. Downsampling randomly removes reads so that the given approximate coverage is obtained. If quality trimming is selected, downsampling is done on the trimmed reads. By default downsampling to 180x coverage is enabeled. However, downsampling can be disabled or the coverage can be changed if wished.
SKESA Options
The number of threads and maximum memory usage can be specified here. Furthermore, additional command line parameters for SKESA can be defined here (see SKESA manual).
If SKESA was used in a pipeline, then the logging output of SKESA can later be viewed in the Procedure tab of the finished Sample.
SKESA Version
By default, the SKESA version in the subdirectory ext/ of the SeqSphere+ Client installation directory is used (version 2.3.0). In the Advanced Settings of SKESA in the pipeline script an alternative version can be defined.
However, SeqSphere+ is only tested with the SKESA version that is delivered with the software. SKESA may be updated with future SeqSphere+ updates.