

Contents
Introduction
The authors from NCBI of the new SKESA de novo assembler claim in their publication (citation) that the assembler
- produces assemblies of equal or better quality than SPAdes,
- is at least 4x faster than SPAdes,
- produces identical results regardless of the number of threads or memory,
- scales well with increase in compute resources (minimum 16 GB RAM recommended) or coverage, and
- handles low-level contamination in reads (contiguity decrease when contamination level increases to 9x or above).
In this evaluation we aim to confirm their claims with a specific focus on allele calling efficiency.
Methods
Three strains with finished genomes were re-sequenced on an Illumina MiSeq machine. The strains cover the whole range of G/C content, i.e., low Staphylococcus aureus strain COL (NC_002951; 2.8 MBases genome), medium Escherichia coli strain Sakai (NC_009089; 5.5 MBases genome), and high Pseudomonas aeruginosa strain PAO1 (NC_002516; 6.3 MBases genome). 250bp Nextera XT paired-end (PE) libraries were produced for all 3 strains. In addition were for S. aureus 150bp and 300bp PE libraries constructed. Finally, two 250bp PE libraries of mixtures with different concentrations of S. aureus and Enterococcus faecium strain ATCC BAA-472 (NC_017960.1; 3 MBases genome) were produced. Assembly of the produced data was performed with the three different de novo assemblers that are available in SeqSphere+: SKESA (version 2.3), SPAdes (version 3.11) and Velvet (version 1.1). Before assembling the data were downsampled to different estimated coverages. After assembly an allele calling was done with SeqSphere+ using cgMLST reference(seed)-only schemes based on the NCBI GenBank entry of the same strain.
The following parameters were compared for different coverages and assemblers:
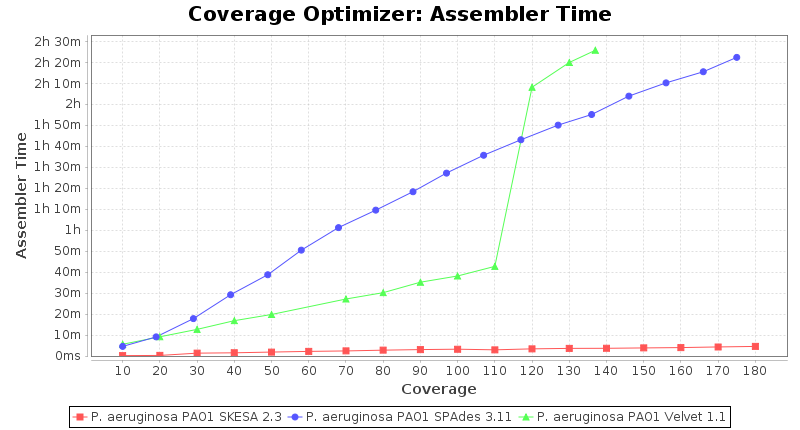
- Assembler Time
- The time the assembling step took within the pipeline (smaller values are better) on an Intel Xeon system with 20 cores and 192 GB memory.
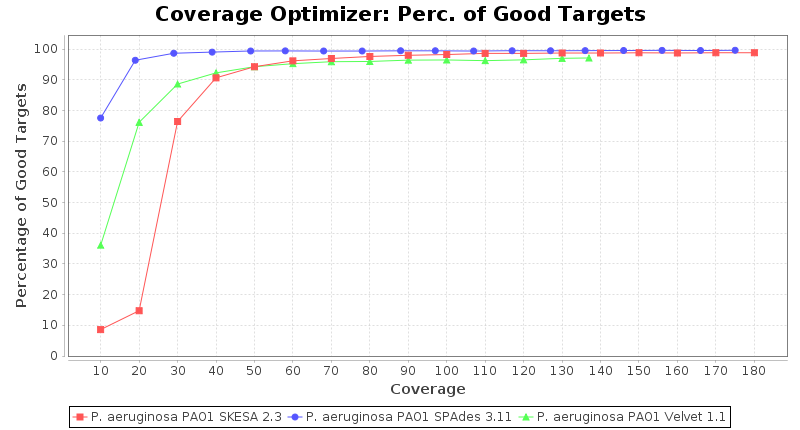
- Percentage of Good cgMLST Targets
- The number of targets that pass the quality check in a reference-only cgMLST task template that was defined with the downloaded NCBI genome of this strain (larger values are better).
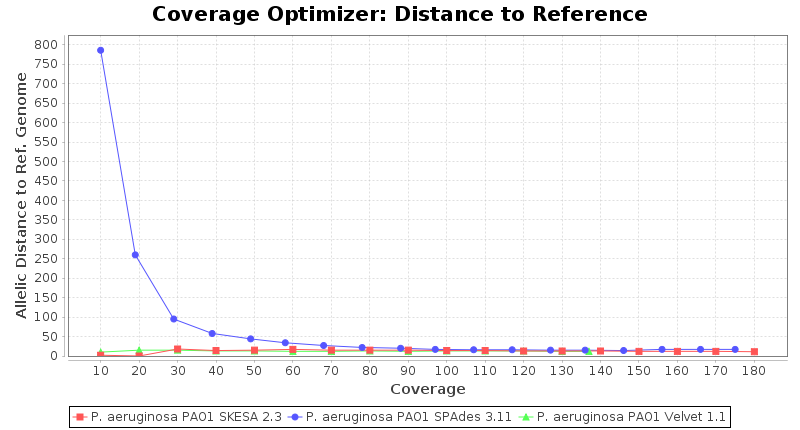
- Allelic Distance to Reference (Seed) Genome
- The absolute number of differing alleles between the assembly and the downloaded NCBI genome of this strain (smaller values are better, but will never be zero due to sequencing errors in the finished NCBI genome and/or micro-evolutionary changes of the culture collection strain during multiple sub-cultivation passages).
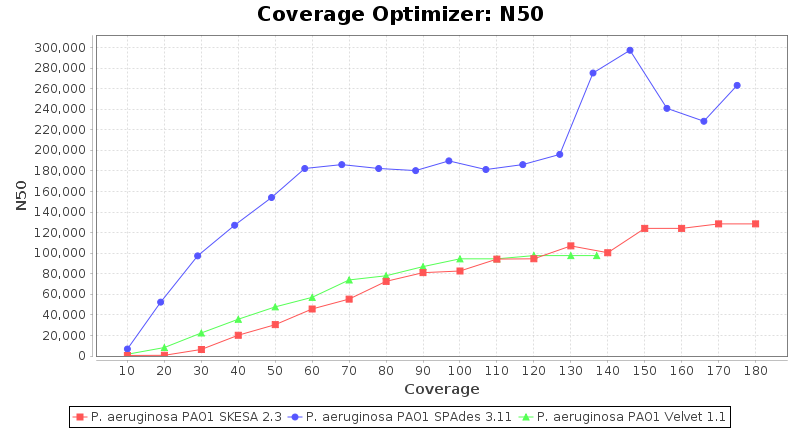
- N50
- The N50 value is a measure for assembly quality in terms of contiguity. N50 is described as a weighted median statistic such that 50% of the entire assembly is contained in contigs or scaffolds equal to or larger than this value.
Results
Assembler Time / Allele Calling Efficiency / N50 from Pure Culture
Staphylococcus aureus 250bp PE
SeqSphere+ used a cgMLST reference-only scheme with 2,486 targets. SKESA and SPAdes were run on Linux whereas Velvet was run on Windows.
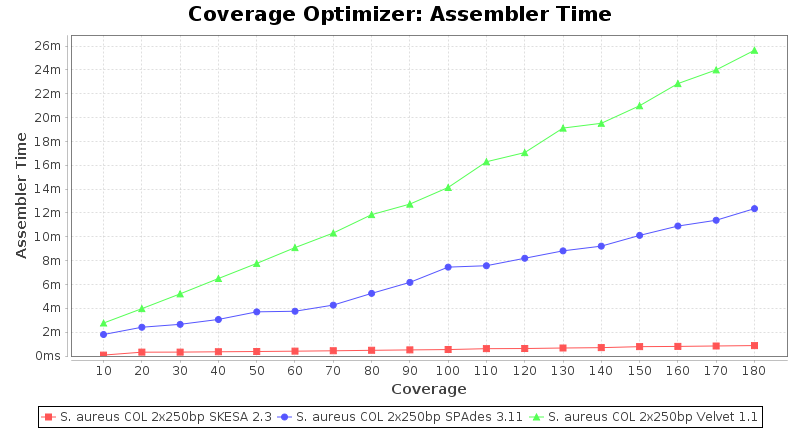
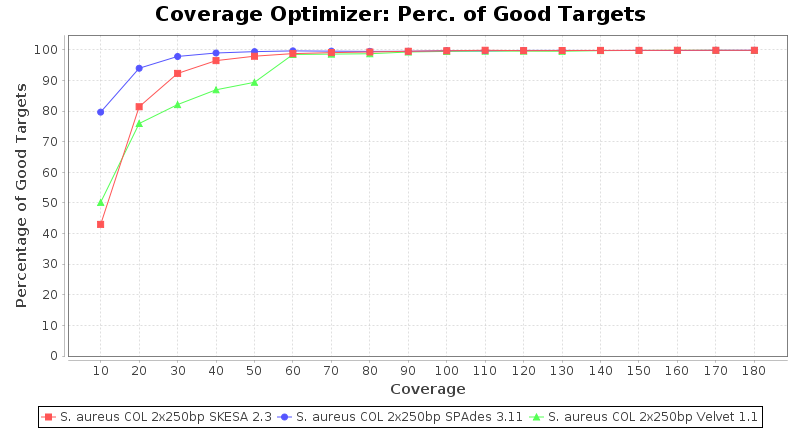
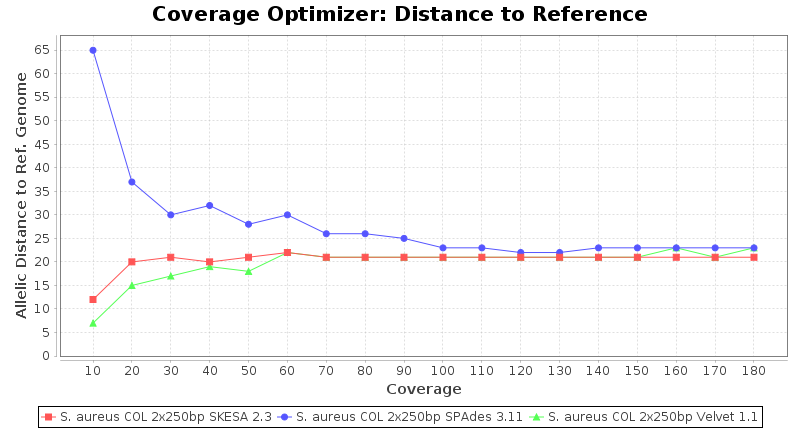
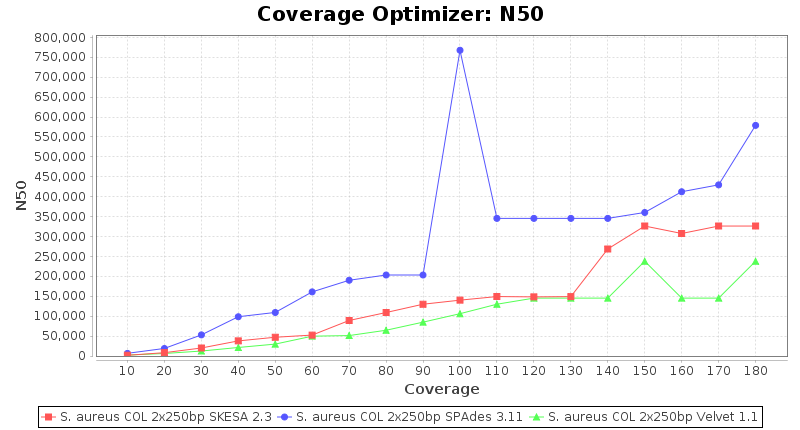
Staphylococcus aureus 150bp PE
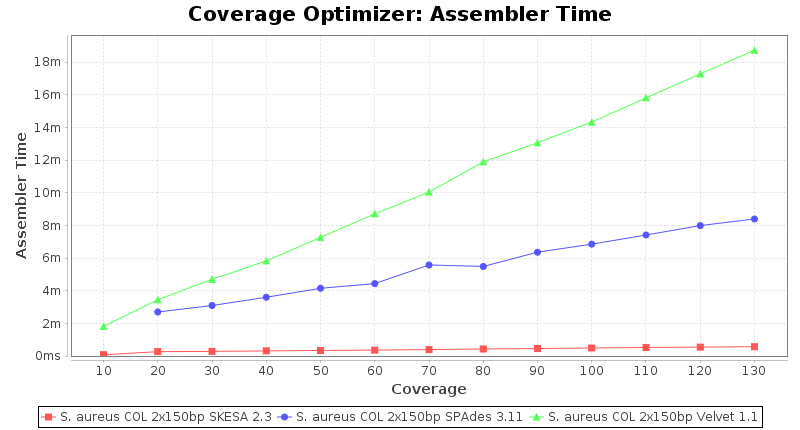
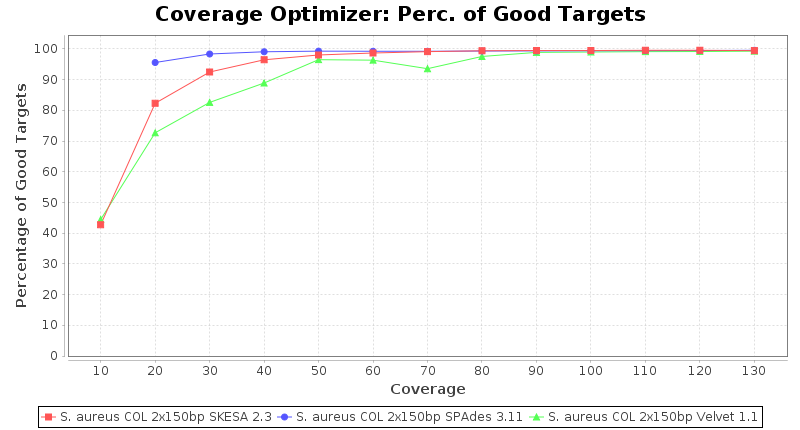
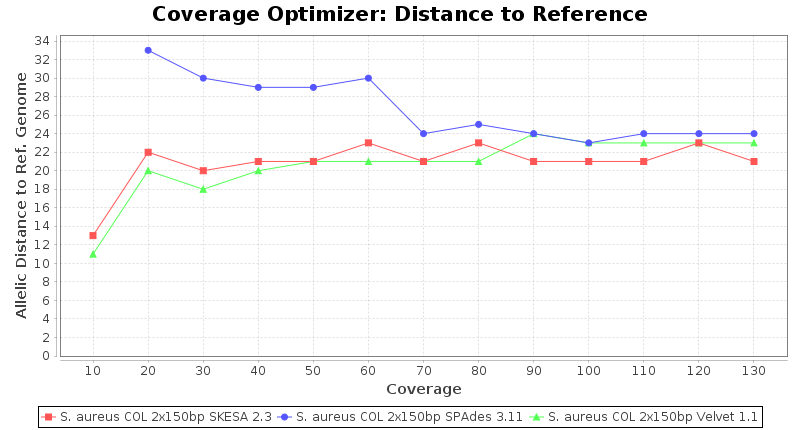
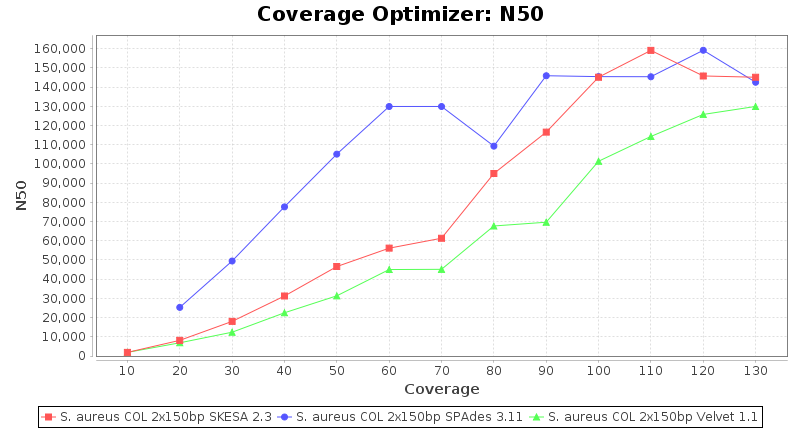
Staphylococcus aureus 300bp PE
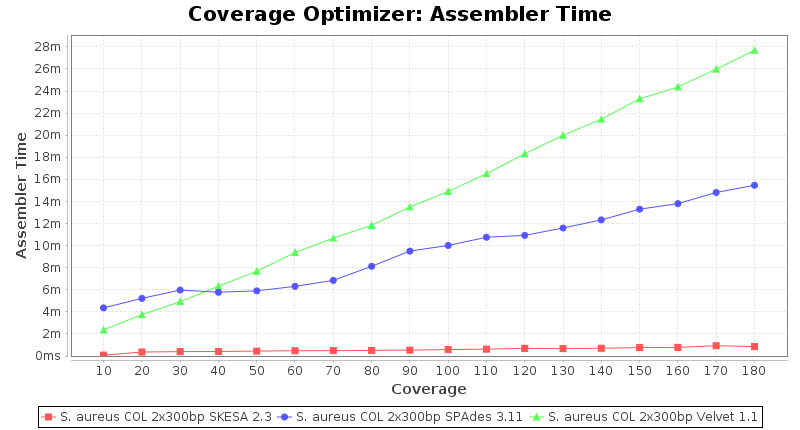
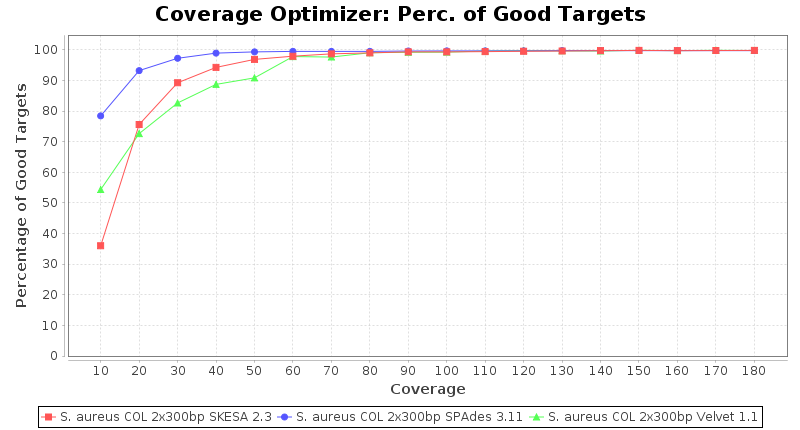
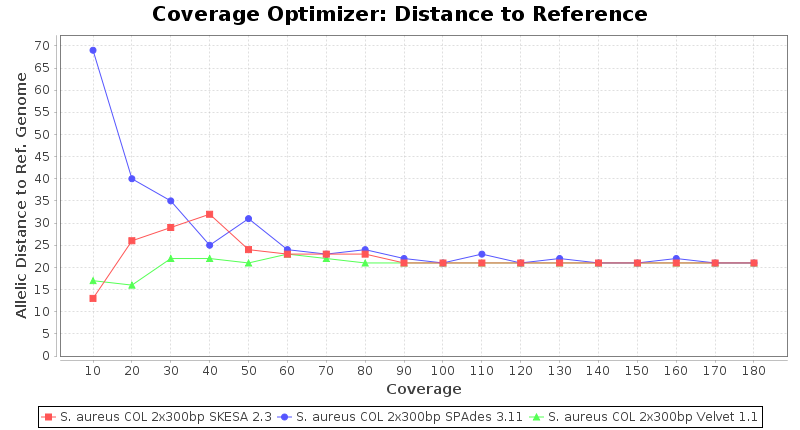
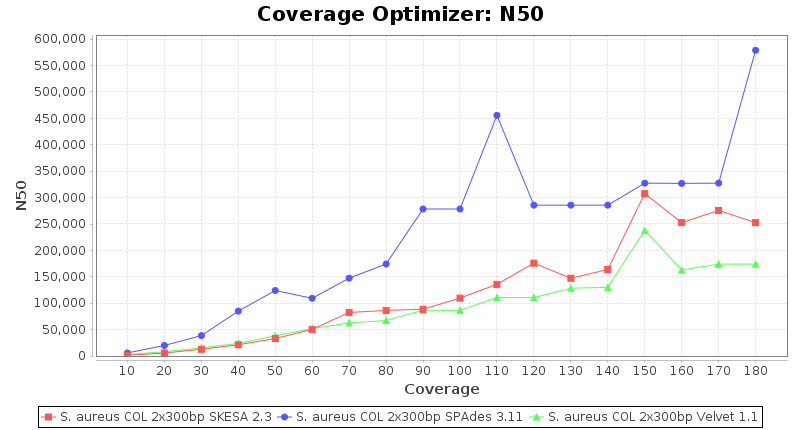
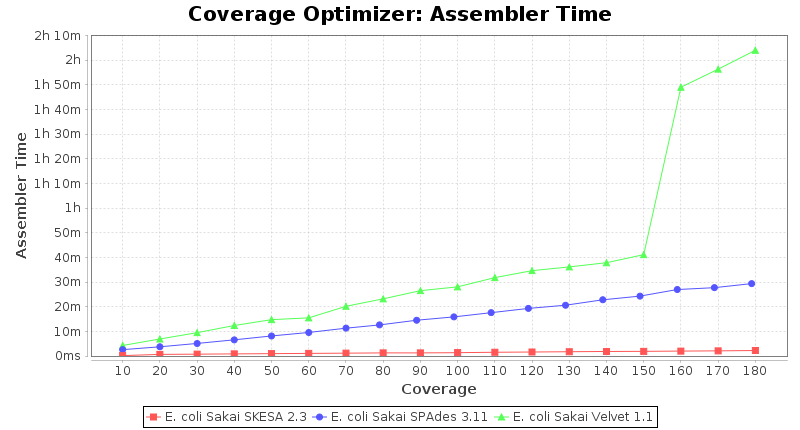
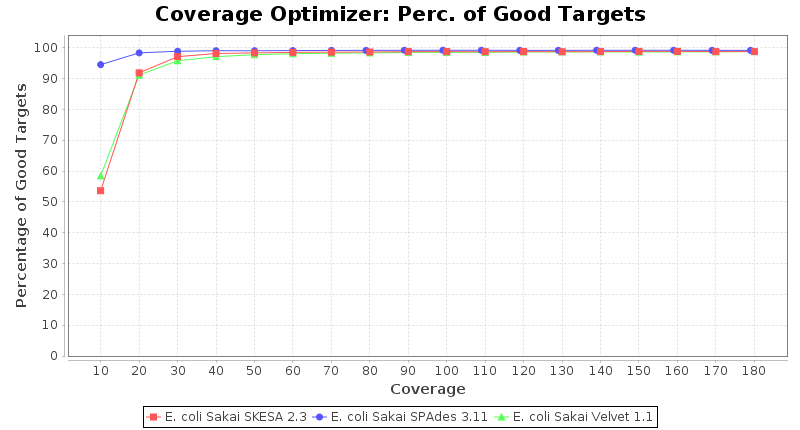
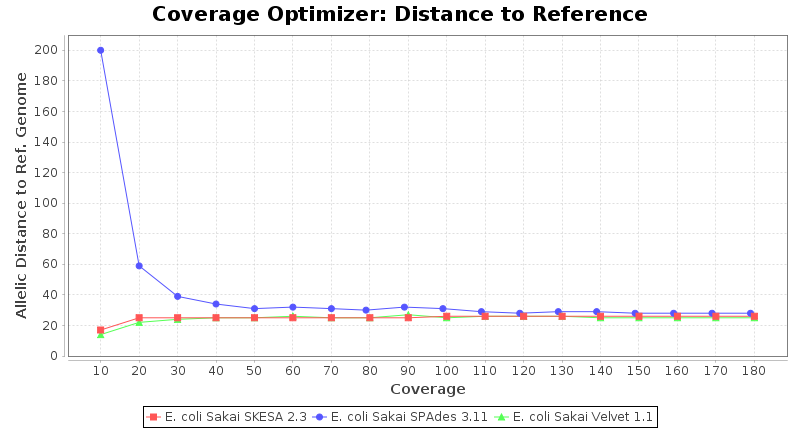
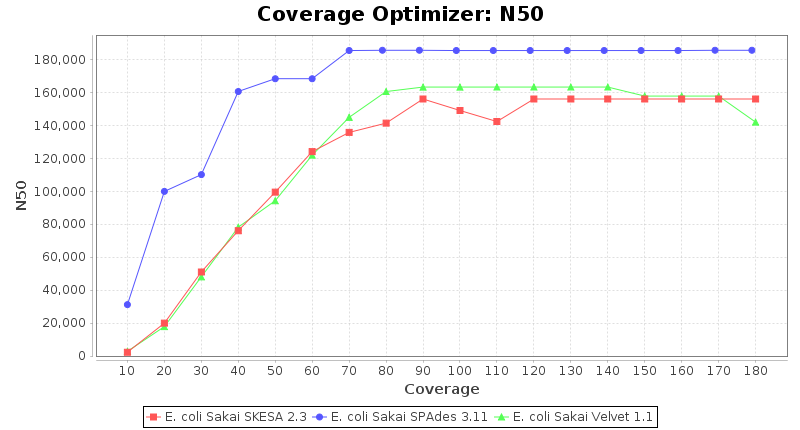
Escherichia coli 250bp PE
SeqSphere+ used a cgMLST reference-only scheme with 4,225 targets. SKESA and SPAdes were run on Linux whereas Velvet was run on Windows.
Pseudomonas aeruginosa 250bp PE
SeqSphere+ used a cgMLST reference-only scheme with 5,267 targets. SKESA and SPAdes were run on Linux whereas Velvet was run on Windows.
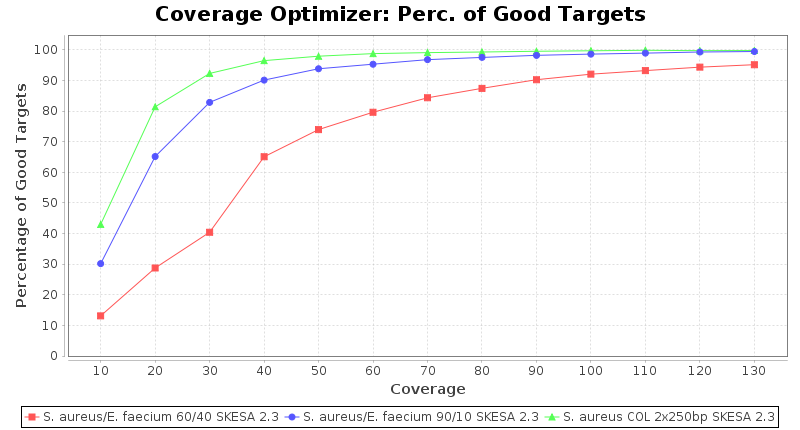
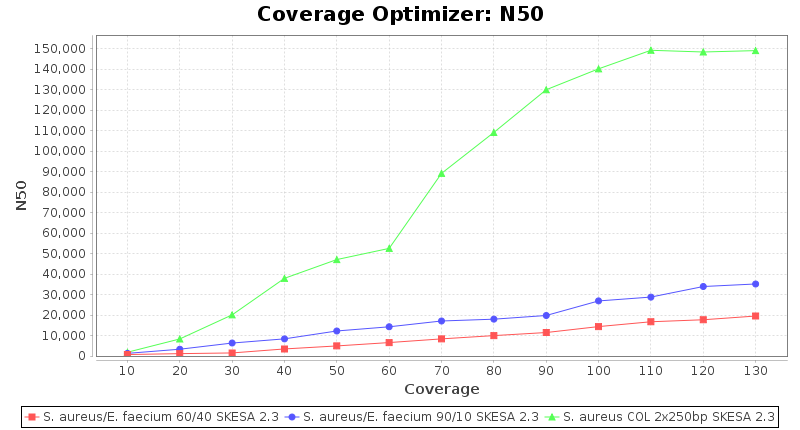
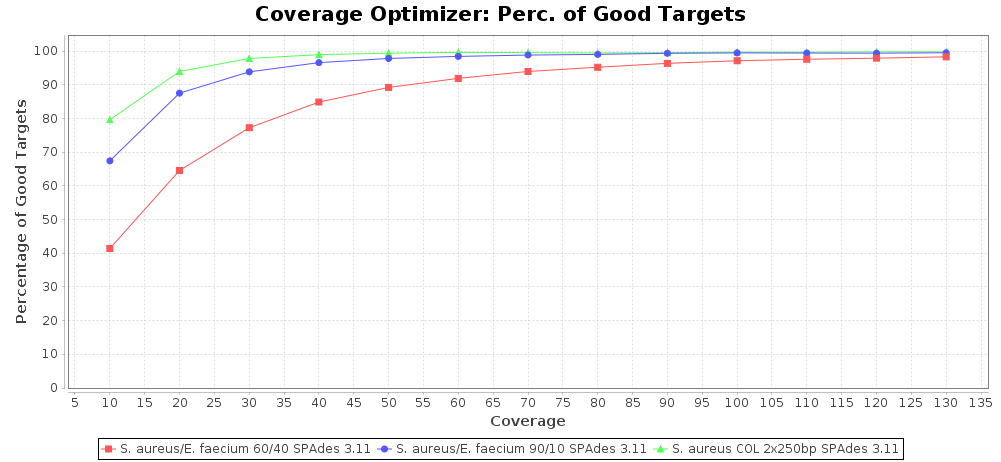
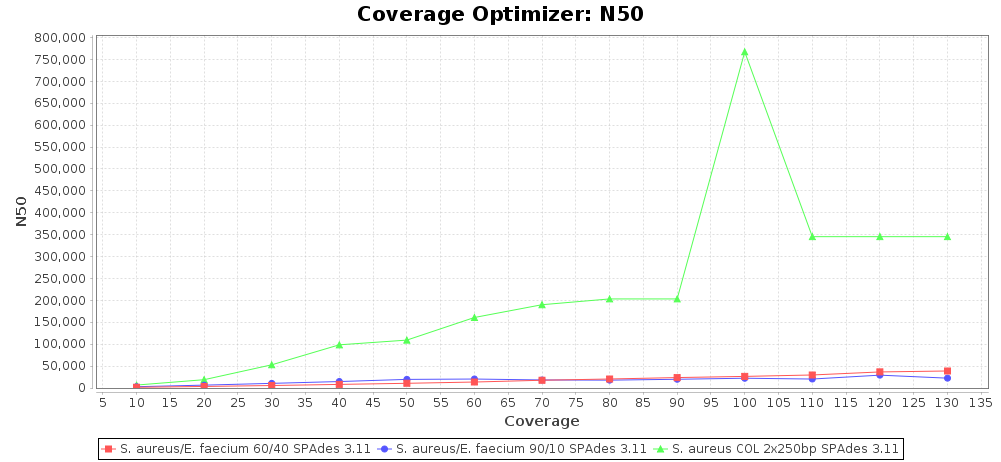
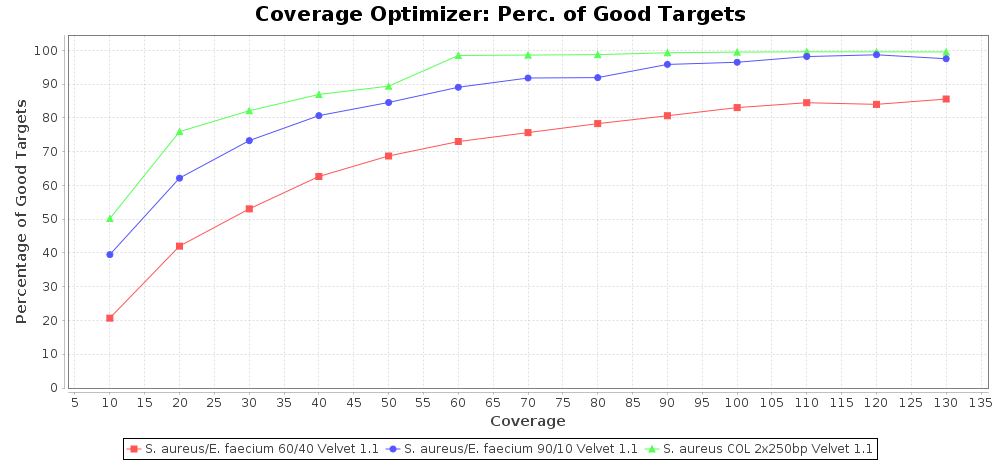
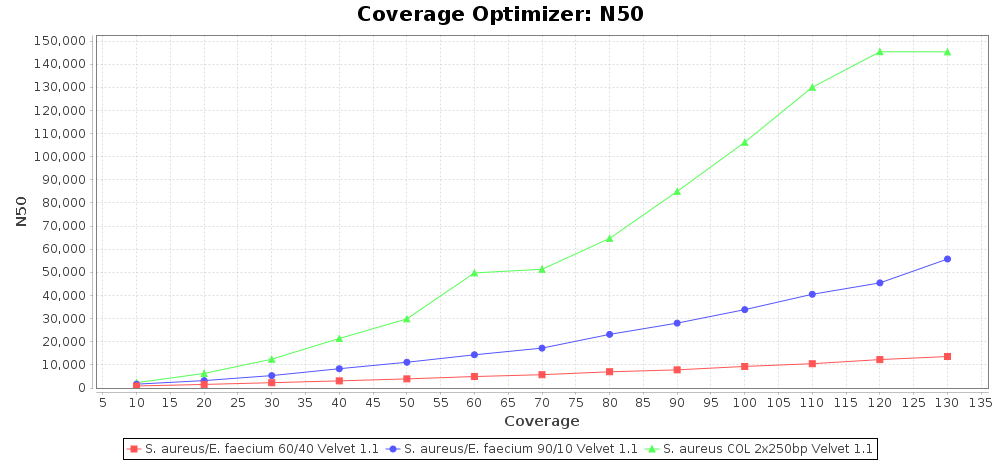
Percentage of Good cgMLST Targets / N50 from Mixed Culture
DNA of S. aureus strain COL (NC_002951; 2.8 MBases genome) and Enterococcus faecium strain ATCC BAA-472 (NC_017960.1; 3 MBases genome) were mixed with 60:40 and 90:10 ratios, respectively. Re-sequencing of 250bp PE Nextera XT libraries was done on a MiSeq. Resulting reads were downsampled to different estimated coverages relative to the COL genome size and processed with SeqSphere+ using a Staphylococcus aureus cgMLST reference-only scheme with 2,486 targets. SKESA and SPAdes were run on Linux, whereas Velvet was run on Windows. For comparison also the pure culture data of the COL strain are shown in the graphs.
SKESA
SPAdes
Velvet
Summary
Those claims of the authors of the SKESA publication that were checked could be verified. SKESA produces indeed very fast high quality de novo assemblies that are very well suited for cgMLST allele calling.