

BWA is a reference genome assisted assembler for read data. It maps reads to a given reference genomes. BWA contains several algorithms, SeqSphere+ supports BWA-SW and BWA-MEM. The BWA-MEM algorithm is recommended as it is much faster than BWA-SW. However, BWA-MEM is only available for Linux, so in SeqSphere+ it is only supported on Linux and on Windows 10 with WSL installed. On Windows without WSL only the older BWA-SW is supported.
BWA is used as part of the SeqSphere+ Assembling Pipeline for remapping reads to de novo assemblies (SKESA/SPAdes) or to perform reference mapping. Reference mapping is only recommended for monomorphic organisms (e.g. M. tuberculosis) as results strongly depend on the similarity of the test draft-genome to the reference. Therefore a warning is shown, if BWA reference mapping is selected in a pipeline for non-monomorphic organisms with public cgMLST schemes.
BWA can also be directly accessed using the menu Tools | Read Mapper (BWA). The reference mapper opens FASTQ-files that contain either single or paired reads. The input files can be quality trimmed and downsampled before mapping. The mapped reads and the reference sequence are stored in a BAM-file.
Contents
Quality Trimming
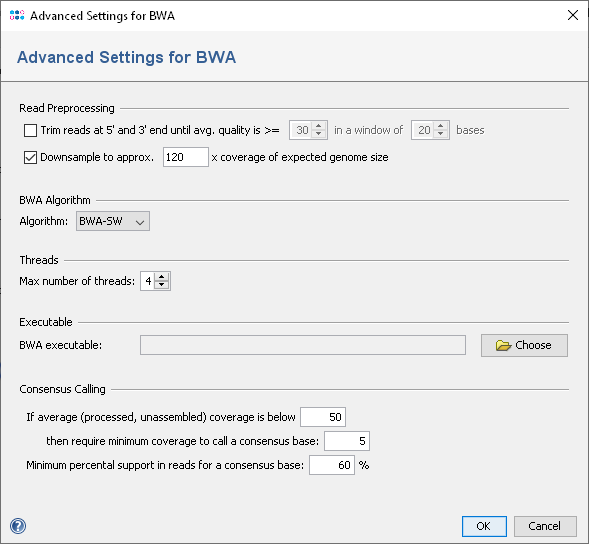
The reads can be processed before they are mapped. They can be automatically trimmed based on read quality and downsampled. BWA does its own trimming, so quality trimming is disabled by default. When trimming is enabled, reads are trimmed on both ends until the average base quality is better than the given value in a window of a selected number of bases.
Downsampling
To reduce the size of the output files and the time required for mapping, the input files can be downsampled. Downsampling randomly removes reads so that the given approx. size is obtained. If quality trimming is selected, downsampling is done on the trimmed reads. Depending on sequencing technology and read length a different downsampling coverage setting might be useful. For Illumina HiSeq/MiSeq data a downsampling to approx. 120 times the expected genome size worked well.
Standard Settings (in pipeline and manual mode)
- Quality trimming: reads can be trimmed from both ends until their quality is above the given average quality within a given window.
- Downsampling: select which coverage of expected genome size should be reached by downsampling.
- Algorithm: select if BWA-SW or BWA-MEM algorithm should be used (default is BWA-MEM if available, else BWA-SW).
- Threads: define the maximum number threads that should be used by bwa.
- BWA Executable: define the path to the bwa executable.
- Consensus Calling: define the settings for the consensus caller postprocessing.
Manual Settings (in manual mode only)
- Reference genome: the genome that is the base for mapping. A FASTA or a GENBANK-file can be used as reference.
- Output directory: the directory where the resulting BAM-files will be written to.
- Read Files: the read files, in FASTQ or FASTQ.GZ format. Multiple files can be selected, and they can be automatically grouped by their filename (e.g. forward and reverse reads can be grouped together).
- Files contain paired reads: Check if the files contain paired reads. The mapper uses paired reads if exactly two files are in each file group. Both forward and reverse files must contain the same amount of reads, and read number X in the forward file must correspond to read number X in the reverse file.
- Threads: define the maximum number threads that should be used by bwa.
- Additional Options: specify additional command line options for bwa (see http://bio-bwa.sourceforge.net/bwa.shtml#3).
Postprocessing
Before a BAM file can be scanned by SeqSphere+, a consensus sequence is automatically calculated by the built in consensus caller. As BWA is known to create inacurrate mappings for low coverage data, the consensus calling procedure is configured to use a threshold of 5 as minimum coverage to call a non-ambiguous base, if the estimated average coverage (unmapped) is below 50. The required read support for a consensus base is 60%.
Speed and Memory
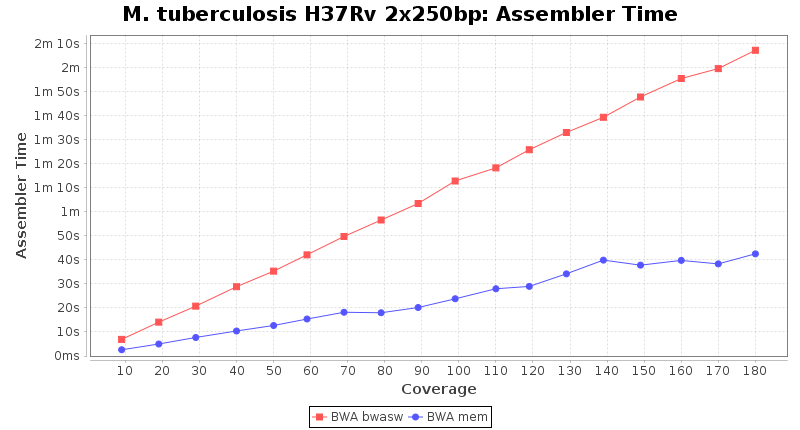
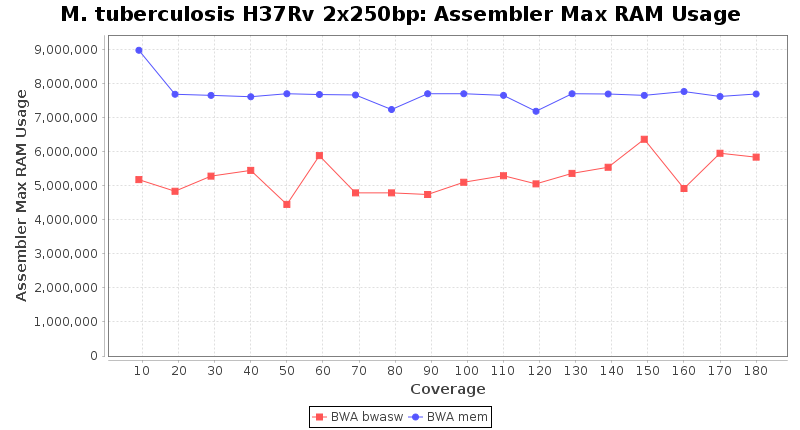
The following charts show the runtime and memory usage for mapping Illumina MiSeq 2x250bp sequence data of Mycobacterium tuberculosis strain H37Rv (NC_000962; 4.4 MBases genome) on an Intel Xeon system with 20 cores and 192 GB memory. Before reference mapping the read files were downsampled to different estimated coverages. The reference mapping was then performed with the two different BWA algorithms BWA-SW (red) and BWA-MEM (blue). In summary, reference mapping does not require huge amounts of memory and is overall much faster than de novo assembly.
Versions and Open Source Note
Only BWA version 0.6 is natively available for Windows, but the BWA-MEM algorithm is only supported since version 0.7. Therefore, SeqSphere+ includes two different versions of BWA: Version 0.6 is used for BWA-SW (on Linux and on any Windows), and version 0.7 is used for BWA-MEM (on Linux and on Windows 10 with WSL installed).
The BWA reference mapping function is a wrapper to an external BWA executable. This BWA executable is open source software that is licensed under the GNU General Public License version 3.0 (GPLv3, http://bio-bwa.sourceforge.net/). The native Windows version of BWA is from https://github.com/xied75/bwa.
For more information on BWA, see: Li H. and Durbin R. (2010) Fast and accurate long-read alignment with Burrows-Wheeler Transform. Bioinformatics, 26: 589-95 [PubMed 20080505] and Li H. (2013) Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv. arXiv:1303.3997 [q-bio.GN] [arXiv].