Contents
Overview
This tutorial describes how to use the Ridom SeqSphere+ software to assembly and analyze bacterial genomic data using the SeqSphere+ Pipeline Mode.
Listeria monocytogenes is used exemplarily for this demonstration. However, by reading this tutorial you should be able to define your own projects for all species with a public available cgMLST scheme in the Task Template Sphere. If you are analyzing a species for which no public cgMLST is available yet, please take a look at Core Genome MLST Schemes help.
Preliminaries
- Installation of SeqSphere+: If SeqSphere+ is not available yet, a one-month trial version can be requested. The SeqSphere+ client and server software can be installed on the same computer for this tutorial.
- System Requirements: This tutorial requires at least 8 GB RAM. It is recommended to use the tutorial on a Windows 10 system with installed Windows Subsystem for Linux (WSL), or on a Linux system. With 8 GB RAM and Core i3 CPU the pipeline takes are around 20 minutes.
 Hint: If a Windows system without WSL is used for the tutorial, the Velvet Assembler must be used as an alternative which increases the runtime of the pipeline fourfold, 16 GB RAM are recommended, and species identification, contamination check, and run details import are not available.
Hint: If a Windows system without WSL is used for the tutorial, the Velvet Assembler must be used as an alternative which increases the runtime of the pipeline fourfold, 16 GB RAM are recommended, and species identification, contamination check, and run details import are not available.
- Tutorial Data: Download the example data archive SeqSphere_Examples_Pipeline_L_monocytogenes_ACCO.zip (~270 MB) for this tutorial, and extract the zip-file on your computer. This example data folder contains Illumina MiSeq 250bp paired-end FASTQ files for 4 isolates of Listeria monocytogenes. The FASTQ files were downsampled to 30x coverage to decrease the assembling time for this tutorial. The original whole genome shotgun (WGS) data was published by Ruppitsch et. al. (JCM 53: 2869–2876, 2015). To demonstrate the import of Illumina run details some artificial run info files were added to the example data folder.
Define Pipeline Script
- Step 1: Start the Ridom SeqSphere+ client without logging in, and press the button
 Start Pipeline Mode on the bottom of the login panel or use the identical menu function in the File menu.
Start Pipeline Mode on the bottom of the login panel or use the identical menu function in the File menu.
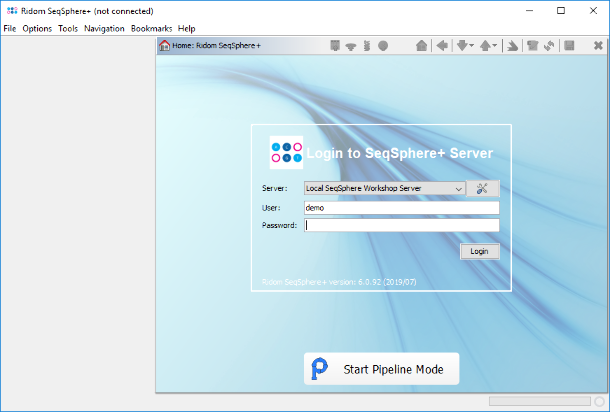
- Step 2: The pipeline mode window starts up. The pipeline mode is designed to run SeqSphere+ in a non-interactive way to assemble, process and analyse WGS data automatically defined by a pipeline script. Press Create New Script to start open a dialog for creating a pipeline script. In the first step the Server Host and the User Login must be defined. Just use localhost for your local computer and the same SeqSphere+ user account that you are normally using for the SeqSphere+ login. The option to store user login in the pipeline script is enabled by default. Below enter the User Password of this user account. If wanted, the password can also be stored (encrypted) in the pipeline script. However, it should be taken into account that if the password is stored in the pipeline script, anyone with access to the computer can run the pipeline. For this tutorial check the Store password encrypted in pipeline script file checkbox.
- Press Next to move on.
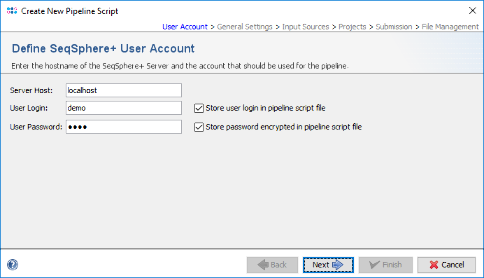
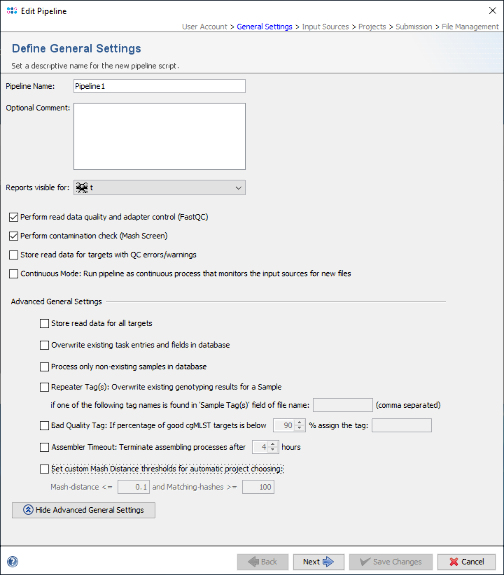
- Step 3: In the Define General Settings panel enter a Pipeline Name (e.g., 'Pipeline Tutorial'). The comment can be left empty and the access control for viewing the reports generated by this pipeline can be left to the user's group as default. If the SeqSphere+ client is running on on Linux or on Windows 10 with installed WSL, four checkbox are shown. The first two checkboxes are preselected and allow to perform automatically a FASTQ file quality control with FastQC and contamination check with Mash. The fourth option, "Continous Mode", can be used to monitor a directory and automatically process newly appearing sequence data files.
- Press Next to move on.
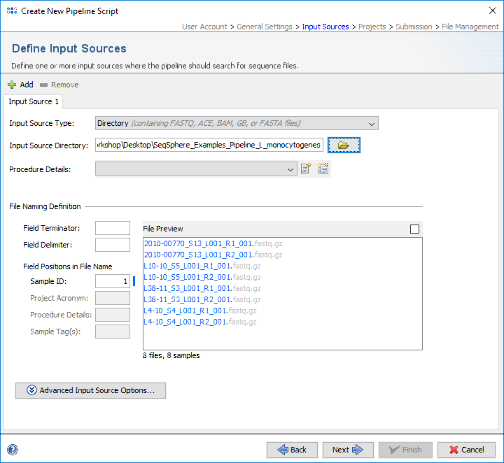
- Step 4: In the next panel the Input Sources for the WGS sequence data are selected. The Input Source Type Directory is predefined. Press the
 button and select the directory SeqSphere_Examples_Pipeline_L_monocytogenes that was unpacked from the downloaded tutorial data file (see Preliminaries). The File Preview on the lower right shows the 8 fastq.gz files that are currently in this directory. When the pipeline is started all files in this directory will be processed. Files can be excluded from processing by the right-click menu in the File Preview.
button and select the directory SeqSphere_Examples_Pipeline_L_monocytogenes that was unpacked from the downloaded tutorial data file (see Preliminaries). The File Preview on the lower right shows the 8 fastq.gz files that are currently in this directory. When the pipeline is started all files in this directory will be processed. Files can be excluded from processing by the right-click menu in the File Preview.
 Important: FASTQ files with adapters (and multiplex indices) trimmed-off are required here for optimal de novo assembly results.
Important: FASTQ files with adapters (and multiplex indices) trimmed-off are required here for optimal de novo assembly results.
- Step 5: The Procedure Details must be selected for the sequence data files. For this tutorial data select Illumina, paired-end reads.
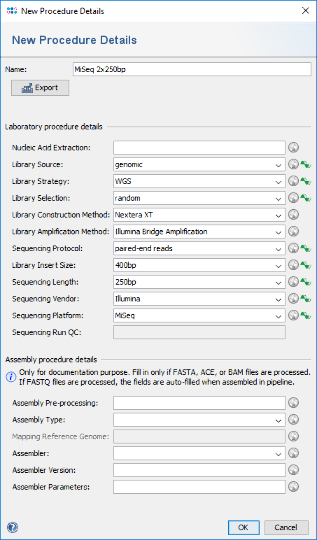
- Hint: Alternatively, for documentary purpose more details can be defined by pushing the
 New Procedure Details button. A New Procedure Details window pops-up. Enter at least Library Source: genomic, Library Strategy: WGS, Library Selection: random, Sequencing Protocol: paired-end reads, Library Insert Size: 400bp, Sequencing Length: 250bp, Sequencing Vendor: Illumina, and Sequencing Platform: MiSeq. The assembly procedure details can be left empty as they are filled-in when using the assembly pipeline. Press the OK button of the New Procedure Details window. Push also the OK button of the Manage Procedure Details window.
New Procedure Details button. A New Procedure Details window pops-up. Enter at least Library Source: genomic, Library Strategy: WGS, Library Selection: random, Sequencing Protocol: paired-end reads, Library Insert Size: 400bp, Sequencing Length: 250bp, Sequencing Vendor: Illumina, and Sequencing Platform: MiSeq. The assembly procedure details can be left empty as they are filled-in when using the assembly pipeline. Press the OK button of the New Procedure Details window. Push also the OK button of the Manage Procedure Details window.
- Step 6: Each input source must also have a File Naming Definition that describes at least how to find the Sample ID in the file names of your sequence data. The Field Terminator is automatically filled with the underline (_) symbol. You can leave this to default for the tutorial data. Press Next to move on.
- Step 7: In the next Define Projects panel the Project(s) are selected into which processed Sample data should be imported. For this tutorial the Project does not exist yet, and must be created. Therefore, press Manage Projects in Database in the bottom right of the window. Press the Create new Project icon to start defining a new project.
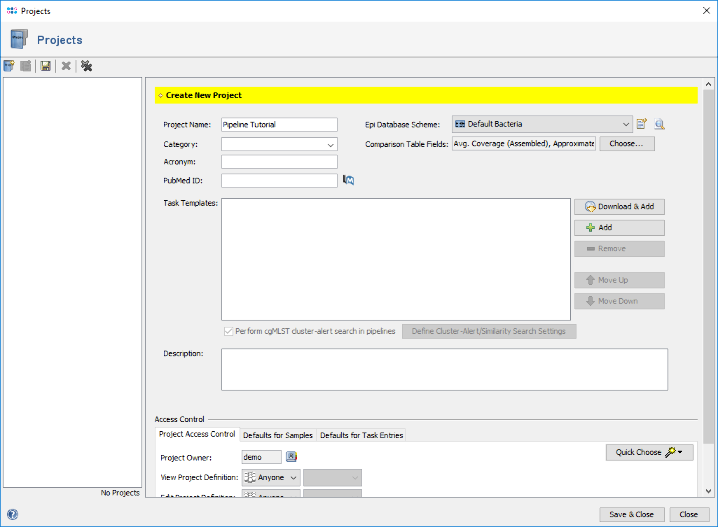
- Step 8: Enter a name for the new Project (e.g., Pipeline Tutorial). Then press
 Download & Add in Task Templates section to browse the Task Template Sphere.
Download & Add in Task Templates section to browse the Task Template Sphere.
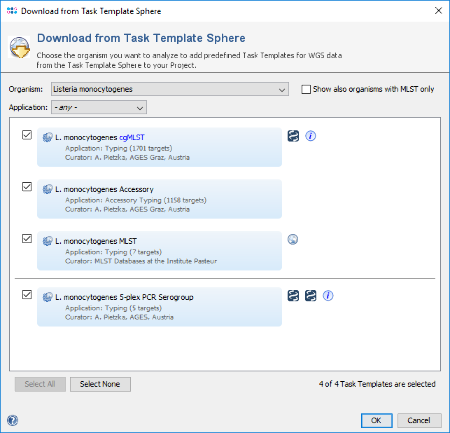
- Step 9: The Task Template Sphere provides all predefined public Task Templates. Choose as organism Listeria monocytogenes. There are four Task Templates available for L. monocytogenes, i.e., cgMLST, Accessory, MLST and 5-plex PCR Serorgroup. The cgMLST Task Template defines the 1,701 genes of the reference strain EDG-e that are used for the public nomenclature and for the definition of the complex type (CT). The Accessory Task Templates defines in addition 1,158 genes that do not belong to the core genome. However, they can be used to increase the discriminatory power if the resolution of cgMLST is not high enough.
- Step 10: Select all four Task Templates and press OK to download and to add them to the Project. Finally confirm with Save & Close to save the new Project.
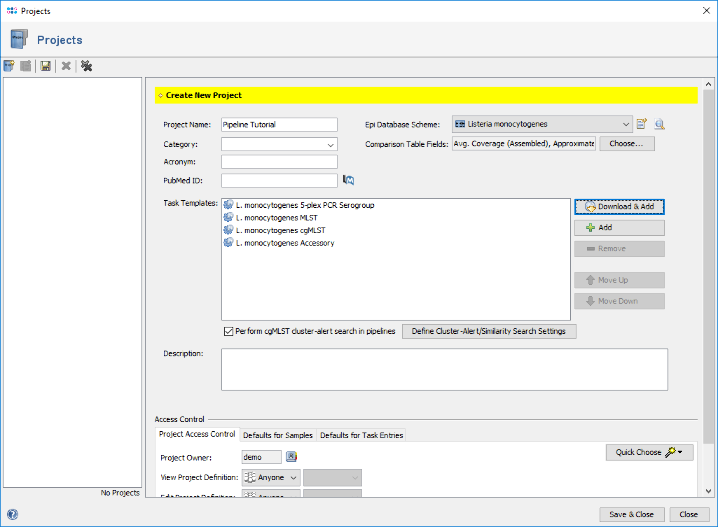
- Step 11: Select the just created project in the Project Name section of the Define Projects panel.
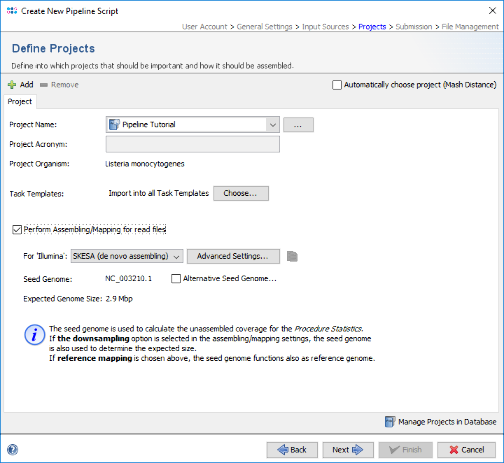
- Step 12: The seed genome for L. monocytogenes that is used as genome size reference for downsampling is automatically loaded from the cgMLST task template in the project. Check the box Perform Assembling/Mapping for read files. If on Linux or Windows 10 with installed WSL the de novo assemlber SKESA is preselected and should be used for this tutorial. Else Velvet can be used for de novo assembling.
- Press Next to move on.
- Step 13: In the upcoming Define Submission panel it can be defined if the pipeline should automatically submit the samples and alleles to the public cgMLST Nomenclature Server. The submission of new alleles is enabled by default and can only be disabled globally in the client. The allelic profile is not stored at cgMLST.org during allele submission. The submission of samples can be used to submit and store the allelic profile and optional metadata on cgMLST.org. In a new pipeline script this is enabled by default, however, it requires a registration of the user at cgMLST.org.
- For this tutorial the option Automatically submit samples to cgMLST.org Nomenclature Server should be unchecked.
- Press Next to move on.
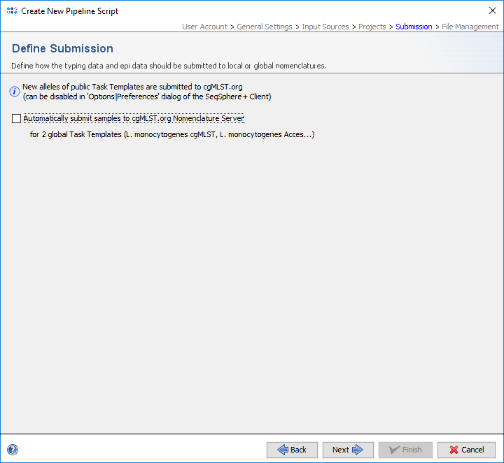
- Step 14: Finally in the Define File Management panel it can be defined what the pipeline should do with the created assembly files and raw reads. Leave all to default and press Test Pipeline Script to validate your pipeline.
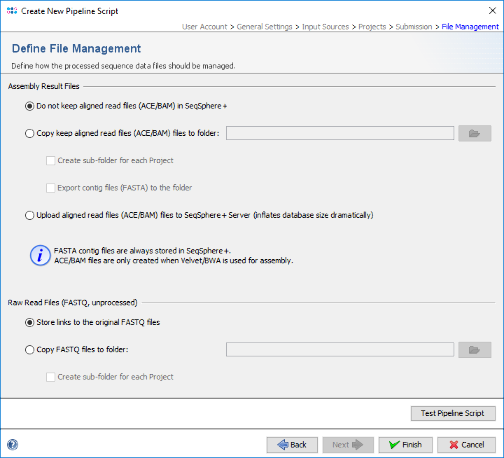
- Step 15: The test should finish successfully. Press Close on this dialog. Now press Finish to store the new pipeline script. Push the button Close of the Manage Pipeline Scripts window.
Run the Pipeline
- Step 1: Be sure that the just created pipeline script is selected and press the button
 Start Script to run the pipeline.
Start Script to run the pipeline.
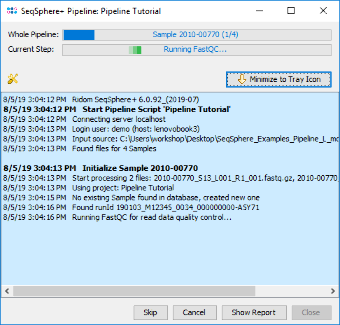
- Step 2: A blue colored progress window is opened, showing the current progress and messages of the pipeline.
- If SKESA is used, the pipeline may take around 20 minutes (8 GB RAM; Core i3 CPU). If Velvet is used, the runtime is quadrupled.
- Step 3: When the pipeline has finished the background color turns to white. Press the Show Report button to see a quick overview for the statistics of the processed Samples.
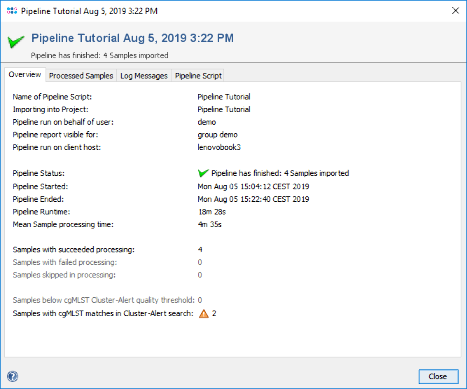
- Step 4: Close the report window, press Close in the pipeline progress window, and exit the pipeline mode with the button Exit and Restart SeqSphere+.
Open the Processed Samples
- Step 1: The SeqSphere+ Client login window appears again. To see further details about the pipeline run and the imported Samples you can now switch back to the normal interactive login session mode. Enter the user name and password and press Login.
- Step 2: On the right of the home screen in the section Recent Pipeline Reports an item for the new report is shown. Click it to open the report, or use the menu function
 Options | Browse Pipeline Reports to choose it from a list of all reports.
Options | Browse Pipeline Reports to choose it from a list of all reports.
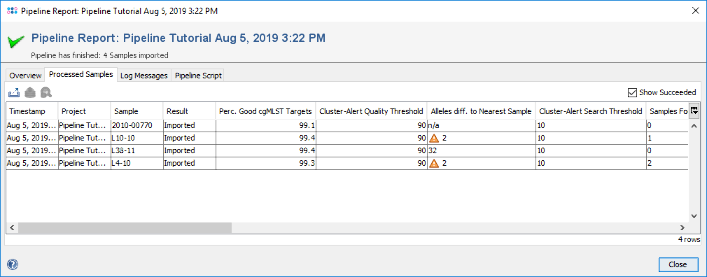
- Step 3: The pipeline report is the same as it was shown just before in the pipeline mode. As SeqSphere+ is now in interactive mode, Samples can be directly loaded into the workspace. Go to the Processed Samples section and double-click on the first sample to load it in the background. Close the pipeline report windows. The Sample is shown in the workspace.
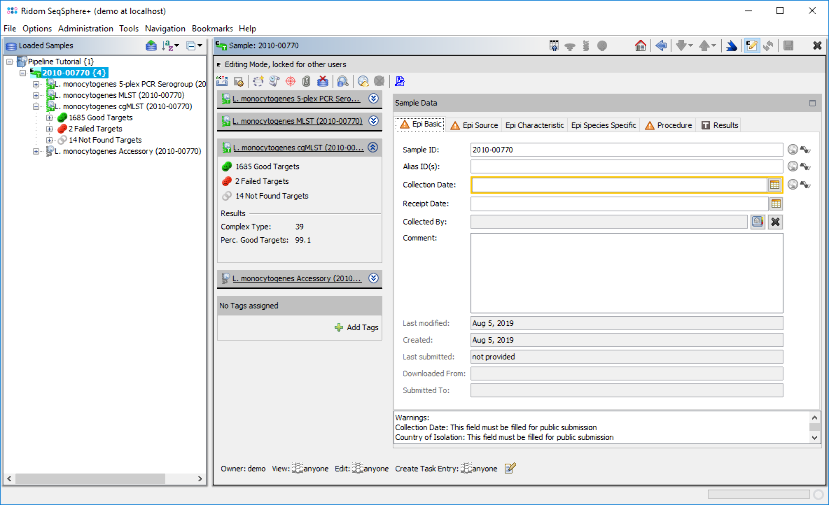
- Step 4: The left panel of the main window shows navigation tree with the loaded Sample. Each Sample node in the navigation has four sub nodes: The 5-plex PCR Serogroup task, the MLST task, the cgMLST task and the Accessory task. Below the task nodes there are the target nodes. Each target node represents one sequence (here a gene) that was extracted from the input data (here the de novo assembled WGS data). The targets can have different target QC states:
-
 Good Targets (green) were extracted and fulfilled all requirements that are defined in the Target QC Procedure of the Task Template.
Good Targets (green) were extracted and fulfilled all requirements that are defined in the Target QC Procedure of the Task Template.
-
 Failed Targets (red) were extracted, but failed at least in one of the requirements that are defined in those parameters. For example, they may have frame shifts and incorrect lengths compared to the allele of the seed genome.
Failed Targets (red) were extracted, but failed at least in one of the requirements that are defined in those parameters. For example, they may have frame shifts and incorrect lengths compared to the allele of the seed genome.
-
 Not Found Targets (gray) were not extracted (because the match did not reached the thresholds in the previous step)
Not Found Targets (gray) were not extracted (because the match did not reached the thresholds in the previous step)
-
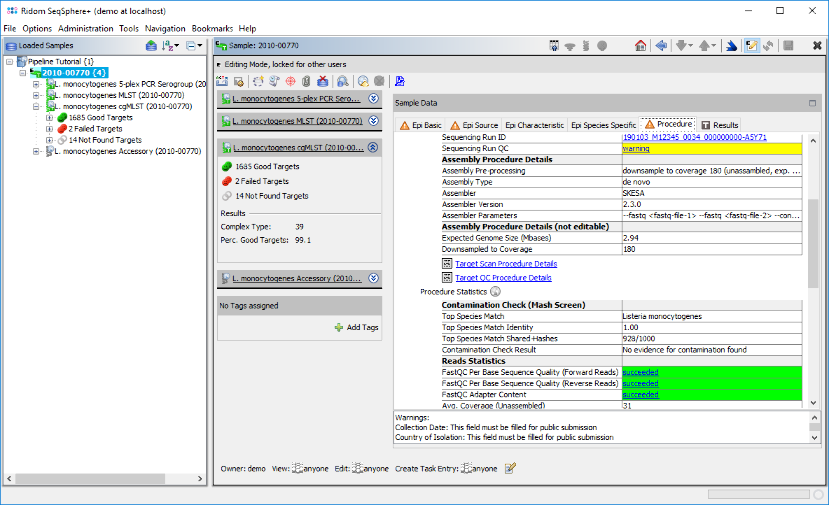
- Step 5: Click on the Procedure tab in the right panel of the window to see the details about the sequence data and processing. Some fields are important for quality control if the sequence data, their QC result status is color highlighted as succeed, warning, or failed.
- In the Procedure Details" section the values for Sequencing Run ID and or Sequencing Run QC can be clicked to show the Sequencing Run Details that were imported by the pipeline.
- In the Reads Statistics section below, the values for etailed of FASTQC Per Base Sequencing Quality and FASTQC Adapter Content can be clicked to show the detailed FastQC results.
- Step 6: Close the sample by pressing the
 in the toolbar above the panel.
in the toolbar above the panel.
Import Epidemiological Metadata
- Step 1: Invoke in the menu File | Import Epi Metadata
- Step 2: In the upcoming Choose File to Import dialog select the file Lm_metadata.xls from example data directory SeqSphere_Examples_Pipeline_L_monocytogenes.
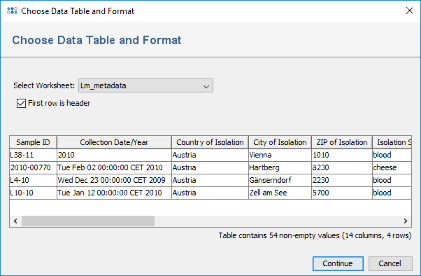
- Step 3: A preview dialog with the content of this Excel file is shown. It contains epidemiological metadata of the four samples for which the sequence data was already processed. Press Continue.
- Step 4: The next dialog defines the import settings. First select on the the the project that was created and processed by the pipeline before.
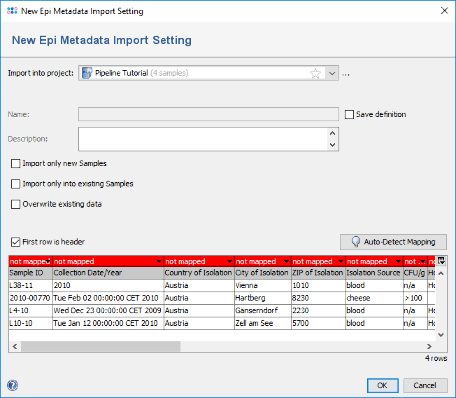
- Step 5: The table on the bottom shows the mapping between the table columns and the SeqSphere+ database fields. By default all columns are unmapped and are therefore highlighted in red. As the column headers have the same (or similar) namings as the database fields, a mapping can be done automatically. Press the button Auto-Detect Mapping to recognize and map all known column names.
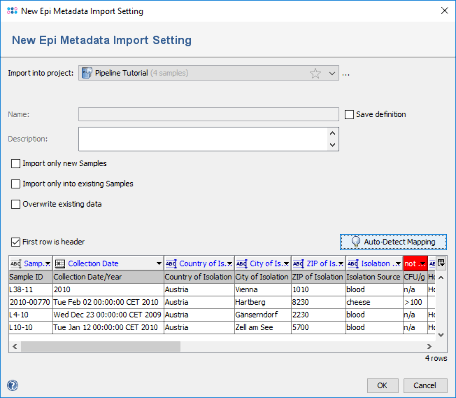
- Step 6: All but two columns were mapped to SeqSphere+ database fields. For the two columns that are still red (CFU/g and Outcome) no fields exist in SeqSphere. If they should be imported in the database, they could be manually mapped to fields by clicking on the red header and selecting a field. If they should be imported into a new field, this field must be created before invoking the metadata import. For this tutorial we leave the two fields unmapped. Press OK to start the import.
- Step 7: After the import is finished, a dialog is shown. Press Open Samples to open the samples and take a short look at the imported database fields (e.g., Collection Date and City of Isolation)
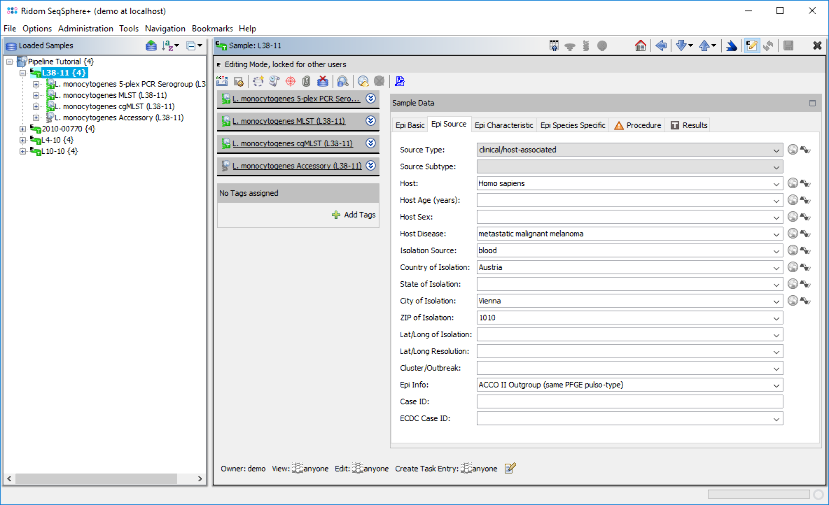
- Step 8: Close all samples by choosing in the menu File | Close All
Analyze Samples with Comparison Table
- Step 1: Choose from the menu Tools | Comparison Table to perform phylogenetic analysis.
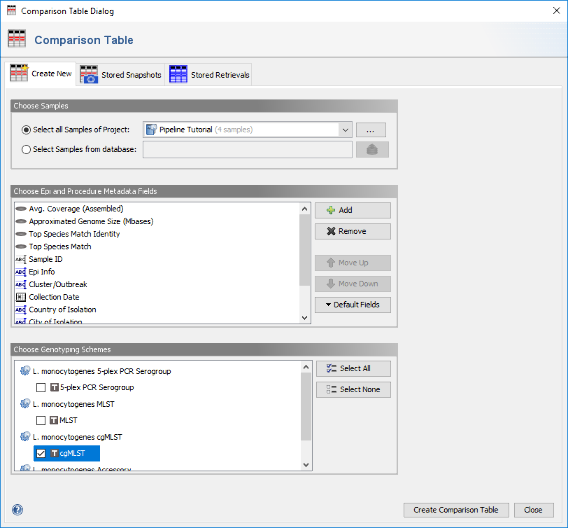
- Step 2: In the Comparison Table dialog go to the first tab "Create New". In the Choose Samples section the new previously created project (e.g., Pipeline Tutorial). Below the default epi metadata fields for a comparison table are listed. On the bottom in the section Choose Genotypings Schemes the checkbox for L. monocytogenes cgMLST is preselected. Press the Create Comparison Table button to confirm the dialog and create the comparison table.
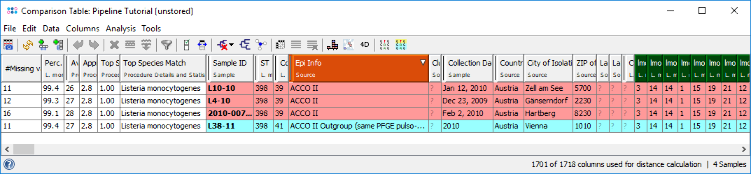
- Step 3: The comparison table is opened and shows the data for the four samples. The column with the red header (Epi Info) is used by default for coloring the sample rows. The columns with a dark green header are used for distance calculation. Those columns are the allele types of the cgMLST task. Some of those contain missing values if a cgMLST target was not found at all in an input sequence ("? (not found)"), or if the Target QC Procedure for this target has failed, e.g., because of a frame shift error ("? (failed)"). The first column in the table shows number of missing values per row.
- Step 4: Right-click on the column Complex Type (seventh column) and choose from the menu
 Set Color Groups by Column Values. Leave the upcoming dialog to defaults and confirm with OK. The Sample rows are now colored by the different cgMLST Complex Types.
Set Color Groups by Column Values. Leave the upcoming dialog to defaults and confirm with OK. The Sample rows are now colored by the different cgMLST Complex Types.
- Step 5: Press the
 Minimum Spanning Tree button in the toolbar to calculate the distances between the Samples and draw a minimum spanning tree for them. Because the table contains missing data, it must be confirmed that the missing values are ignored pairwise. Confirm with OK.
Minimum Spanning Tree button in the toolbar to calculate the distances between the Samples and draw a minimum spanning tree for them. Because the table contains missing data, it must be confirmed that the missing values are ignored pairwise. Confirm with OK.
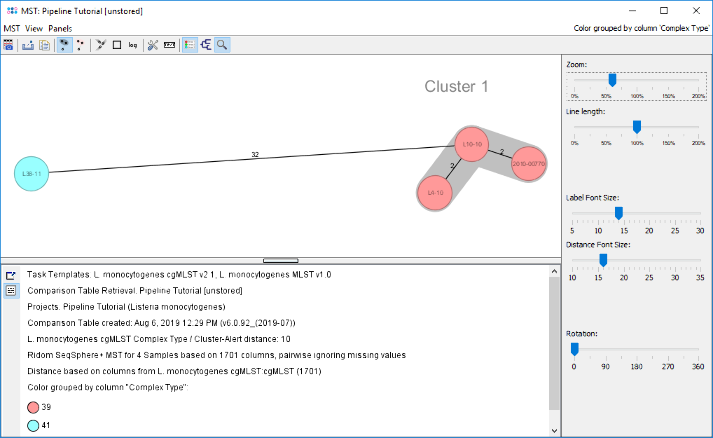
- Step 6: The minimum spanning tree is calculated for the allelic profiles of the 1,701 cgMLST targets (pairwise ignored missing values) and is shown in a new window. By default the nodes are again colored by the Complex Type (CT) and it can be easily seen that 3 of the 4 isolates have the same Complex Type. Just L38-11 belongs to a different Complex Type.
- Two conclusions can be drawn from this tree.
- The three ACCO II isolates have the same Complex Type and a close distance. This indicates epidemiological relationship.
- The fourth isolate (L38-11) has a different Complex Type and a distance of 32 alleles to the three other isolates and does not belong to the outbreak.
- Hint: The MST can be exported by clicking the
 Export MST icon of the toolbar. In the upcoming Export MST file dialog choose as file type the Scalable Vector Graphics (*.svg) or Windows Enhanced Metafile (*.emf) format. Note that EMF or SVG are vector graphics formats and therefore suited for finishing publication ready figures. EMF files can imported and scaled by MS PowerPoint. SVG files can be edited, e.g., with Adobe Illustrator or the open-source InkScape tool (once the file is loaded first ungroup all objects).
Export MST icon of the toolbar. In the upcoming Export MST file dialog choose as file type the Scalable Vector Graphics (*.svg) or Windows Enhanced Metafile (*.emf) format. Note that EMF or SVG are vector graphics formats and therefore suited for finishing publication ready figures. EMF files can imported and scaled by MS PowerPoint. SVG files can be edited, e.g., with Adobe Illustrator or the open-source InkScape tool (once the file is loaded first ungroup all objects).
- Step 7: Press the button
 to store the comparison table and the MST as Comparison Table Snapshot in the database for later reuse.
to store the comparison table and the MST as Comparison Table Snapshot in the database for later reuse.
- Step 8: Press the button
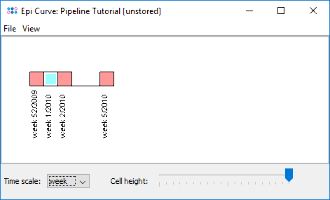
 to create an epi curve for the comparison tables. As the collection dates were imported from the metadata file, the samples are shown in the graph ordered by date. It can be seen that the two outbreak clones appeared successively. The time scale of the epi curve can be changed. samples with imprecise time data are shown with a white rectangle border (e.g., L38-10).
to create an epi curve for the comparison tables. As the collection dates were imported from the metadata file, the samples are shown in the graph ordered by date. It can be seen that the two outbreak clones appeared successively. The time scale of the epi curve can be changed. samples with imprecise time data are shown with a white rectangle border (e.g., L38-10).
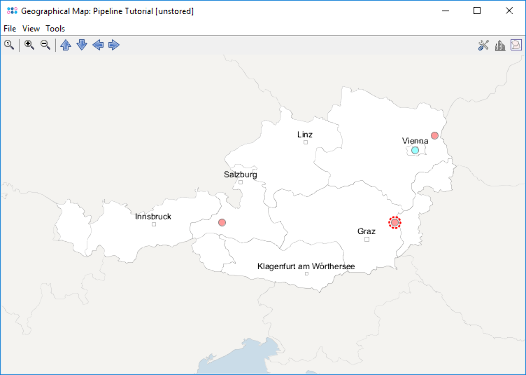
- Step 9: Press the button
 to create an geographical map for the comparison tables. The country and city information was imported from the metadata file but no latitude/longitude information exists yet. Therefore, a dialog is shown that offers to perform a geocoding.
to create an geographical map for the comparison tables. The country and city information was imported from the metadata file but no latitude/longitude information exists yet. Therefore, a dialog is shown that offers to perform a geocoding.
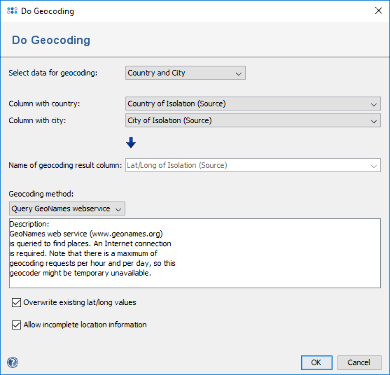
- Step 10: Leave the dialog to default settings and confirm it, to retrieve the geographical latitude and longitude for the city and country information that was imported as metadata. After the data was retrieved, the window with the geographical maps opens automatically. The four samples are drawn on their geographical location. Clicking on a sample in the map marks the sample also in the epi curve, the minimum spanning tree, and in the comparison table.